Parks Donovan H, Imelfort Michael, Skennerton Connor T, Hugenholtz Philip, Tyson Gene W
Australian Centre for Ecogenomics, School of Chemistry and Molecular Biosciences, The University of Queensland, St. Lucia, QLD 4072, Queensland, Australia;
Australian Centre for Ecogenomics, School of Chemistry and Molecular Biosciences, The University of Queensland, St. Lucia, QLD 4072, Queensland, Australia; Institute for Molecular Bioscience, The University of Queensland, St. Lucia, QLD 4072, Queensland, Australia;
Genome Res. 2015 Jul;25(7):1043-55. doi: 10.1101/gr.186072.114. Epub 2015 May 14.
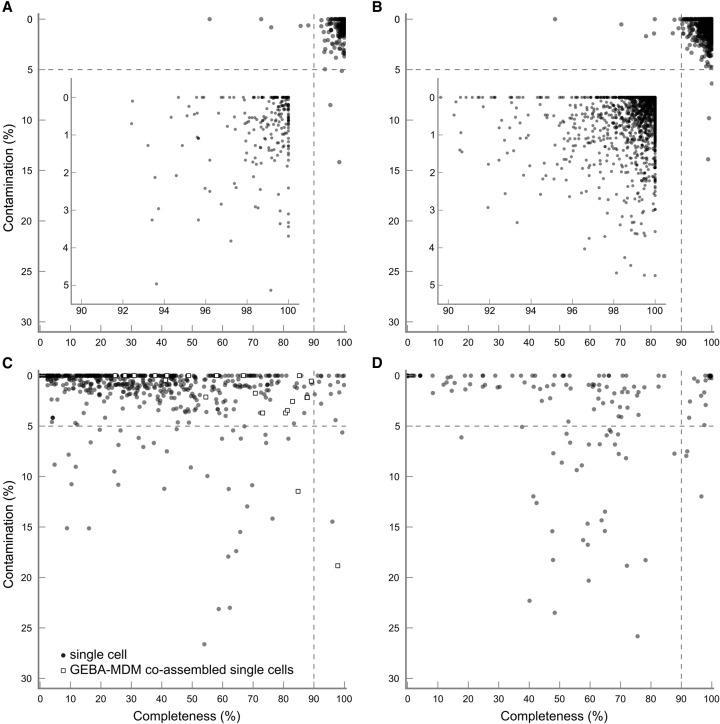
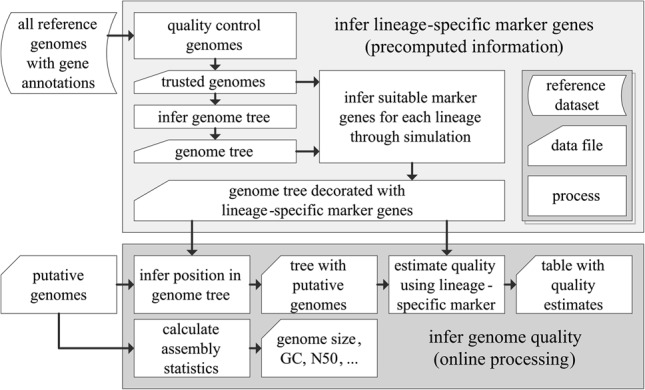
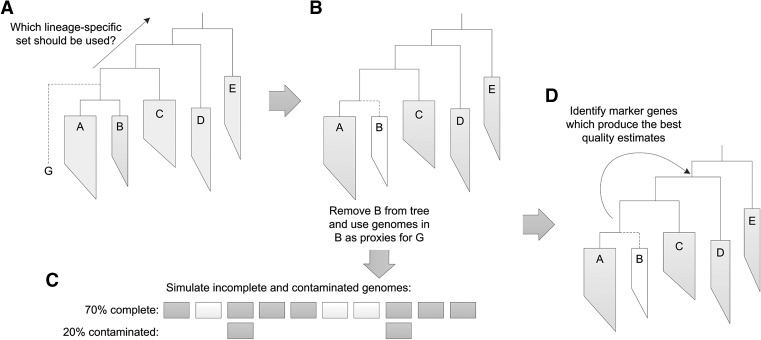
Large-scale recovery of genomes from isolates, single cells, and metagenomic data has been made possible by advances in computational methods and substantial reductions in sequencing costs. Although this increasing breadth of draft genomes is providing key information regarding the evolutionary and functional diversity of microbial life, it has become impractical to finish all available reference genomes. Making robust biological inferences from draft genomes requires accurate estimates of their completeness and contamination. Current methods for assessing genome quality are ad hoc and generally make use of a limited number of "marker" genes conserved across all bacterial or archaeal genomes. Here we introduce CheckM, an automated method for assessing the quality of a genome using a broader set of marker genes specific to the position of a genome within a reference genome tree and information about the collocation of these genes. We demonstrate the effectiveness of CheckM using synthetic data and a wide range of isolate-, single-cell-, and metagenome-derived genomes. CheckM is shown to provide accurate estimates of genome completeness and contamination and to outperform existing approaches. Using CheckM, we identify a diverse range of errors currently impacting publicly available isolate genomes and demonstrate that genomes obtained from single cells and metagenomic data vary substantially in quality. In order to facilitate the use of draft genomes, we propose an objective measure of genome quality that can be used to select genomes suitable for specific gene- and genome-centric analyses of microbial communities.
计算方法的进步以及测序成本的大幅降低,使得从分离株、单细胞和宏基因组数据中大规模恢复基因组成为可能。尽管草图基因组的覆盖范围不断扩大,为微生物生命的进化和功能多样性提供了关键信息,但完成所有可用的参考基因组已变得不切实际。从草图基因组中做出可靠的生物学推断需要准确估计其完整性和污染程度。当前评估基因组质量的方法是临时的,通常利用在所有细菌或古细菌基因组中保守的有限数量的“标记”基因。在这里,我们介绍了CheckM,这是一种自动化方法,用于使用更广泛的标记基因集来评估基因组质量,这些标记基因特定于参考基因组树中基因组的位置以及这些基因的排列信息。我们使用合成数据以及各种源自分离株、单细胞和宏基因组的基因组,证明了CheckM的有效性。结果表明,CheckM能够准确估计基因组的完整性和污染程度,并且优于现有方法。使用CheckM,我们识别出了目前影响公开可用分离株基因组的各种错误,并证明从单细胞和宏基因组数据获得的基因组在质量上存在很大差异。为了便于使用草图基因组,我们提出了一种基因组质量的客观衡量标准,可用于选择适合微生物群落特定基因和基因组分析的基因组。