Maphis Nicole, Xu Guixiang, Kokiko-Cochran Olga N, Cardona Astrid E, Ransohoff Richard M, Lamb Bruce T, Bhaskar Kiran
Department of Molecular Genetics and Microbiology, University of New Mexico Albuquerque, NM, USA.
Department of Neurosciences, Cleveland Clinic Foundation Cleveland, OH, USA.
Front Neurosci. 2015 Jun 3;9:196. doi: 10.3389/fnins.2015.00196. eCollection 2015.
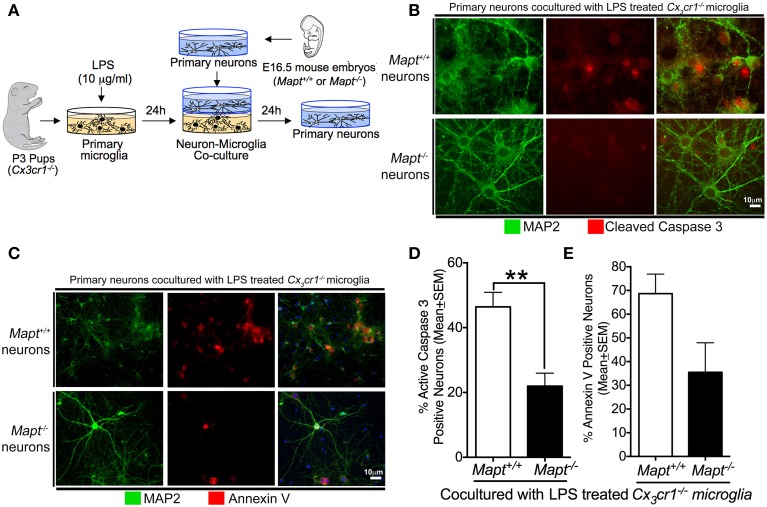
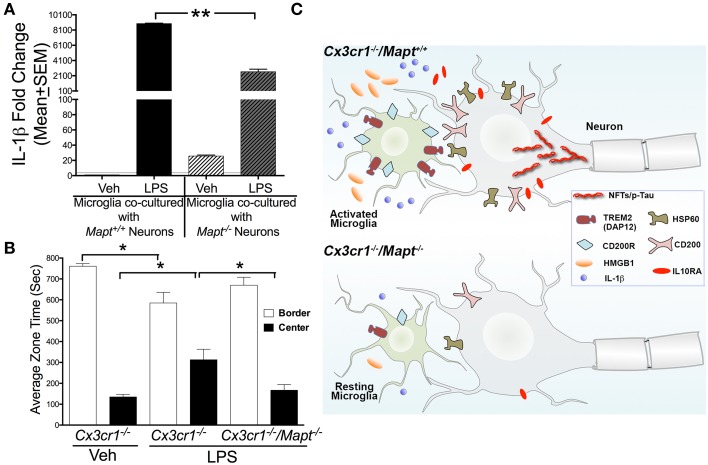
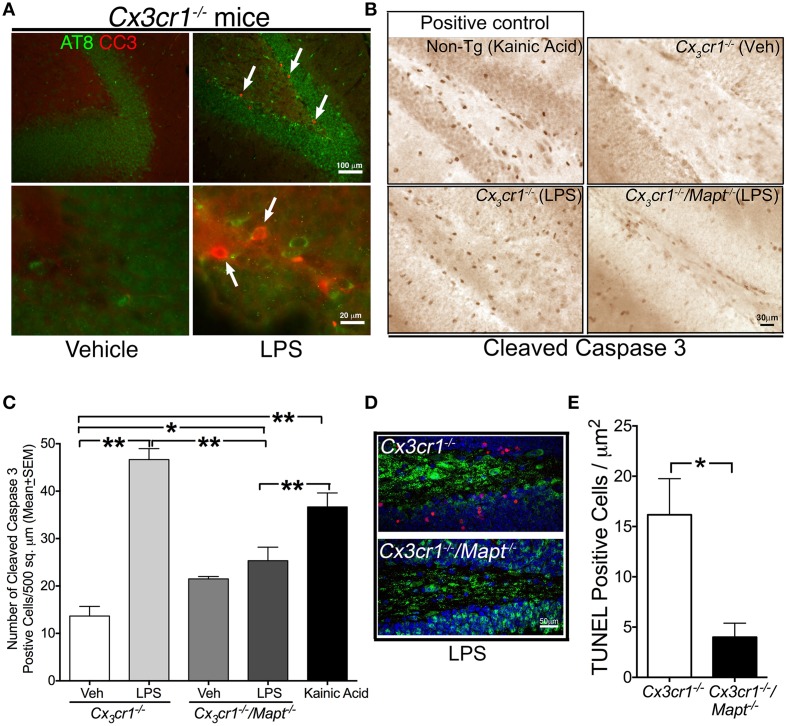
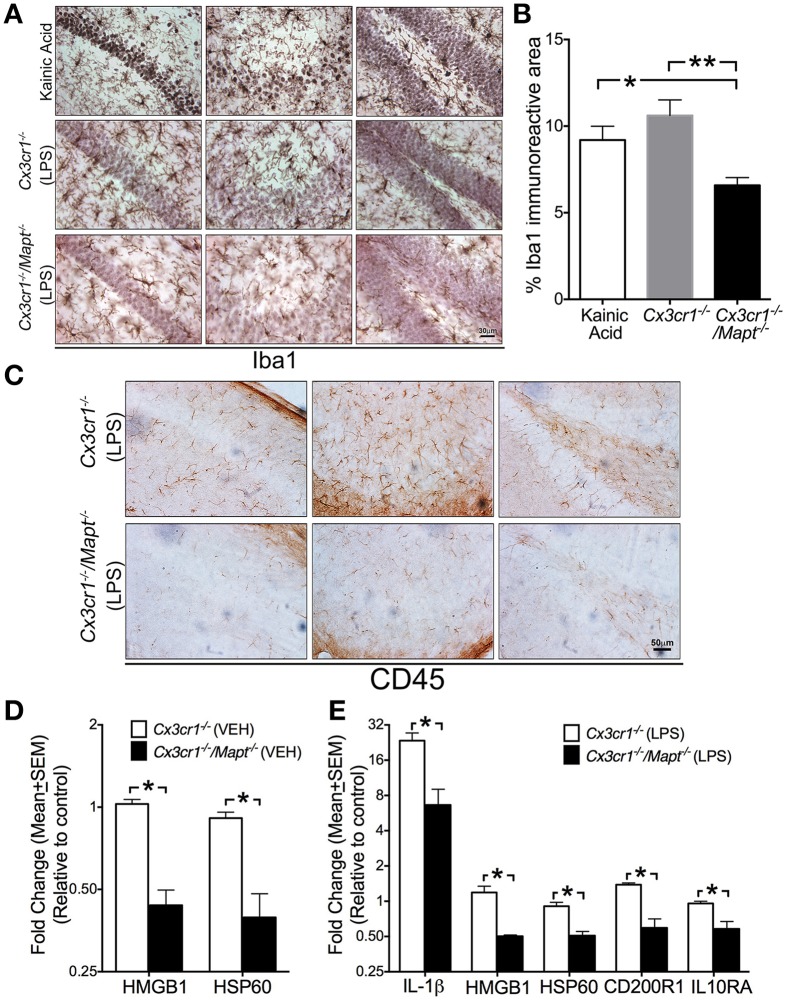
Neuroinflammation is one of the neuropathological hallmarks of Alzheimer's disease (AD) and related tauopathies. Activated microglia spatially coexist with microtubule-associated protein tau (Mapt or tau)-burdened neurons in the brains of human AD and non-AD tauopathies. Numerous studies have suggested that neuroinflammation precedes tau pathology and that induction or blockage of neuroinflammation via lipopolysaccharide (LPS) or anti-inflammatory compounds (such as FK506) accelerate or block tau pathology, respectively in several animal models of tauopathy. We have previously demonstrated that microglia-mediated neuroinflammation via deficiency of the microglia-specific chemokine (fractalkine) receptor, CX3CR1, promotes tau pathology and neurodegeneration in a mouse model of LPS-induced systemic inflammation. Here, we demonstrate that tau mediates the neurotoxic effects of LPS in Cx3cr1 (-/-) mice. First, Mapt (+/+) neurons displayed elevated levels of Annexin V (A5) and TUNEL (markers of neurodegeneration) when co-cultured with LPS-treated Cx3cr1 (-/-)microglia, which is rescued in Mapt (-/-) neurons. Second, a neuronal population positive for phospho-S199 (AT8) tau in the dentate gyrus is also positive for activated or cleaved caspase (CC3) in the LPS-treated Cx3cr1 (-/-) mice. Third, genetic deficiency for tau in Cx3cr1 (-/-) mice resulted in reduced microglial activation, altered expression of inflammatory genes and a significant reduction in the number of neurons positive for CC3 compared to Cx3cr1 (-/-)mice. Finally, Cx3cr1 (-/-)mice exposed to LPS displayed a lack of inhibition in an open field exploratory behavioral test, which is rescued by tau deficiency. Taken together, our results suggest that pathological alterations in tau mediate inflammation-induced neurotoxicity and that deficiency of Mapt is neuroprotective. Thus, therapeutic approaches toward either reducing tau levels or blocking neuroinflammatory pathways may serve as a potential strategy in treating tauopathies.
神经炎症是阿尔茨海默病(AD)及相关tau蛋白病的神经病理学特征之一。在人类AD和非AD tau蛋白病患者的大脑中,活化的小胶质细胞与微管相关蛋白tau(Mapt或tau)负荷过重的神经元在空间上共存。大量研究表明,神经炎症先于tau蛋白病变,并且在几种tau蛋白病动物模型中,通过脂多糖(LPS)诱导或抗炎化合物(如FK506)阻断神经炎症,分别会加速或阻断tau蛋白病变。我们之前已经证明,在脂多糖诱导的全身炎症小鼠模型中,小胶质细胞特异性趋化因子( fractalkine)受体CX3CR1缺乏介导的小胶质细胞神经炎症会促进tau蛋白病变和神经退行性变。在此,我们证明tau蛋白介导了LPS对Cx3cr1(-/-)小鼠的神经毒性作用。首先,当与LPS处理的Cx3cr1(-/-)小胶质细胞共培养时,Mapt(+/+)神经元的膜联蛋白V(A5)和TUNEL(神经退行性变标志物)水平升高,而在Mapt(-/-)神经元中这种情况得到缓解。其次,在LPS处理的Cx3cr1(-/-)小鼠中,齿状回中磷酸化S199(AT8)tau阳性的神经元群体,其活化或裂解的半胱天冬酶(CC3)也呈阳性。第三,与Cx3cr1(-/-)小鼠相比,Cx3cr1(-/-)小鼠中tau蛋白的基因缺陷导致小胶质细胞活化减少、炎症基因表达改变,且CC3阳性神经元数量显著减少。最后,暴露于LPS的Cx3cr1(-/-)小鼠在旷场探索行为测试中表现出缺乏抑制作用,而tau蛋白缺陷可使其得到缓解。综上所述,我们的结果表明,tau蛋白的病理改变介导了炎症诱导的神经毒性,而Mapt缺陷具有神经保护作用。因此,降低tau蛋白水平或阻断神经炎症途径的治疗方法可能是治疗tau蛋白病的潜在策略。