Srilekha Sundaramurthy, Arokiasamy Tharigopala, Srikrupa Natarajan N, Umashankar Vetrivel, Meenakshi Swaminathan, Sen Parveen, Kapur Suman, Soumittra Nagasamy
SNONGC Department of Genetics and Molecular Biology, Vision Research Foundation, Chennai, India; Ph.D Scholar, Birla Institute of Technology & Science (BITS), Hyderabad, India.
SNONGC Department of Genetics and Molecular Biology, Vision Research Foundation, Chennai, India.
PLoS One. 2015 Jul 6;10(7):e0131679. doi: 10.1371/journal.pone.0131679. eCollection 2015.
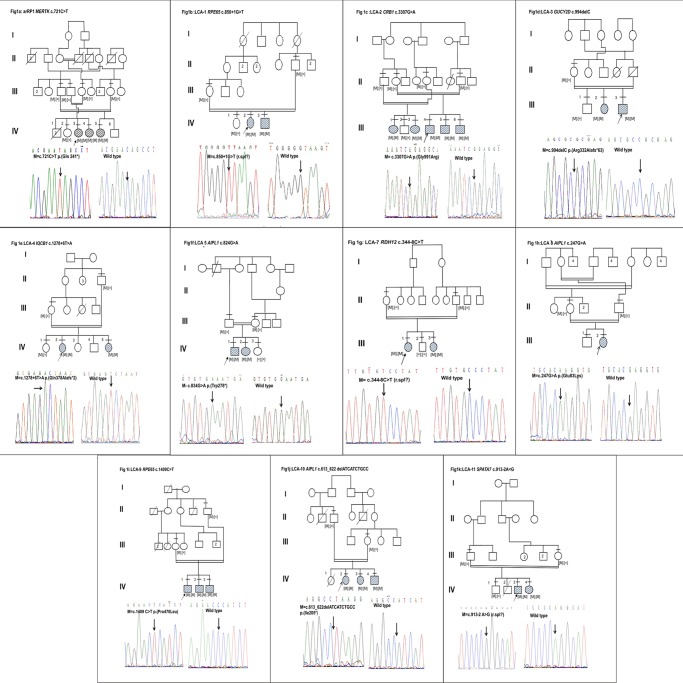
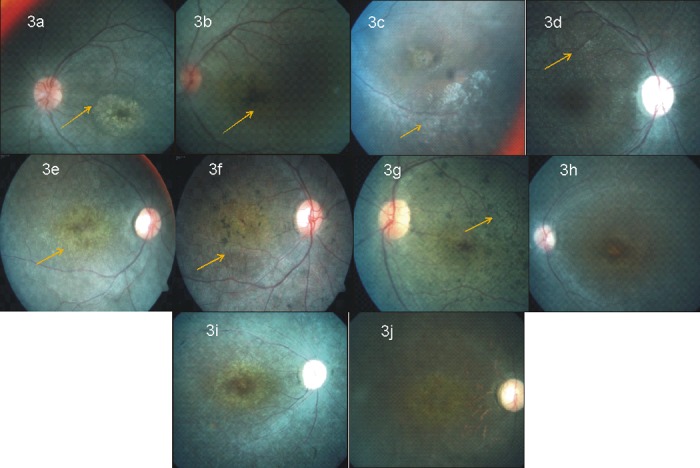
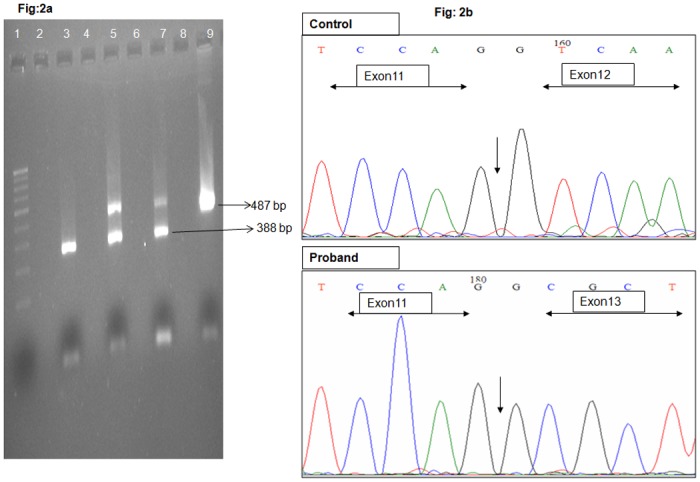
Leber congenital amaurosis (LCA) and retinitis pigmentosa (RP) are retinal degenerative diseases which cause severe retinal dystrophy affecting the photoreceptors. LCA is predominantly inherited as an autosomal recessive trait and contributes to 5% of all retinal dystrophies; whereas RP is inherited by all the Mendelian pattern of inheritance and both are leading causes of visual impairment in children and young adults. Homozygosity mapping is an efficient strategy for mapping both known and novel disease loci in recessive conditions, especially in a consanguineous mating, exploiting the fact that the regions adjacent to the disease locus will also be homozygous by descent in such inbred children. Here we have studied eleven consanguineous LCA and one autosomal recessive RP (arRP) south Indian families to know the prevalence of mutations in known genes and also to know the involvement of novel loci, if any. Complete ophthalmic examination was done for all the affected individuals including electroretinogram, fundus photograph, fundus autofluorescence, and optical coherence tomography. Homozygosity mapping using Affymetrix 250K HMA GeneChip on eleven LCA families followed by screening of candidate gene(s) in the homozygous block identified mutations in ten families; AIPL1 - 3 families, RPE65- 2 families, GUCY2D, CRB1, RDH12, IQCB1 and SPATA7 in one family each, respectively. Six of the ten (60%) mutations identified are novel. Homozygosity mapping using Affymetrix 10K HMA GeneChip on the arRP family identified a novel nonsense mutation in MERTK. The mutations segregated within the family and was absent in 200 control chromosomes screened. In one of the eleven LCA families, the causative gene/mutation was not identified but many homozygous blocks were noted indicating that a possible novel locus/gene might be involved. The genotype and phenotype features, especially the fundus changes for AIPL1, RPE65, CRB1, RDH12 genes were as reported earlier.
莱伯先天性黑蒙(LCA)和视网膜色素变性(RP)是视网膜退行性疾病,可导致严重的视网膜营养不良,影响光感受器。LCA主要以常染色体隐性遗传特征遗传,占所有视网膜营养不良的5%;而RP则通过所有孟德尔遗传模式遗传,两者都是儿童和年轻人视力损害的主要原因。纯合性定位是在隐性疾病中定位已知和新疾病基因座的有效策略,特别是在近亲交配中,利用疾病基因座附近的区域在这种近亲繁殖的儿童中也将通过遗传而纯合的事实。在这里,我们研究了11个南印度近亲LCA家庭和1个常染色体隐性RP(arRP)家庭,以了解已知基因中突变的发生率,并了解是否有新基因座的参与。对所有受影响个体进行了全面的眼科检查,包括视网膜电图、眼底照片、眼底自发荧光和光学相干断层扫描。对11个LCA家庭使用Affymetrix 250K HMA基因芯片进行纯合性定位,随后在纯合子区域筛选候选基因,在10个家庭中鉴定出突变;分别在3个家庭中发现AIPL1突变,2个家庭中发现RPE65突变,1个家庭中分别发现GUCY2D、CRB1、RDH12、IQCB1和SPATA7突变。鉴定出的10个突变中有6个(60%)是新的。对arRP家庭使用Affymetrix 10K HMA基因芯片进行纯合性定位,在MERTK中鉴定出一个新的无义突变。该突变在家族中分离,在筛选的200条对照染色体中未出现。在11个LCA家庭中的一个家庭中,未鉴定出致病基因/突变,但发现了许多纯合子区域,表明可能涉及一个新的基因座/基因。AIPL1、RPE65、CRB1、RDH12基因的基因型和表型特征,特别是眼底变化,如先前报道。