Health Research Institute, Research Center of Thalassemia and Hemoglobinopathy, Ahvaz Jundishapur University of Medical Sciences, Ahvaz, Iran.
Department of Hematology, Faculty of Medical Sciences, Tarbiat Modares University, Tehran, Iran.
Cell J. 2015 Summer;17(2):193-200. doi: 10.22074/cellj.2016.3713. Epub 2015 Jul 11.
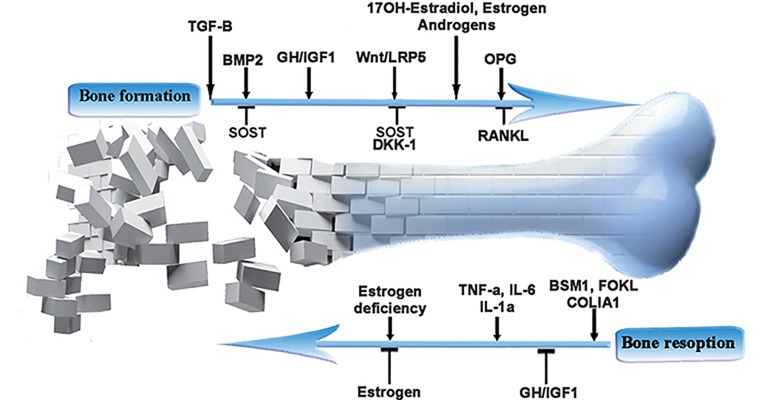
β-thalassemia is the most common single gene disorder worldwide, in which hemoglobin β-chain production is decreased. Today, the life expectancy of thalassemic patients is increased because of a variety of treatment methods; however treatment related complications have also increased. The most common side effect is osteoporosis, which usually occurs in early adulthood as a consequence of increased bone resorption. Increased bone resorption mainly results from factors such as delayed puberty, diabetes mellitus, hypothyroidism, ineffective hematopoiesis as well as hyperplasia of the bone marrow, parathyroid gland dysfunction, toxic effect of iron on osteoblasts, growth hormone (GH) and insulin-like growth factor-1 (IGF-1) deficiency. These factors disrupt the balance between osteoblasts and osteoclasts by interfering with various molecular mechanisms and result in decreased bone density. Given the high prevalence of osteopenia and osteoporosis in thalassemic patients and complexity of their development process, the goal of this review is to evaluate the molecular aspects involved in osteopenia and osteoporosis in thalassemic patients, which may be useful for therapeutic purposes.
β-地中海贫血是全球最常见的单基因疾病,其血红蛋白β链的产生减少。如今,由于各种治疗方法的应用,地中海贫血患者的预期寿命得以延长,但与之相关的治疗并发症也有所增加。最常见的副作用是骨质疏松症,这通常发生在成年早期,是由于骨吸收增加所致。骨吸收增加主要是由于青春期延迟、糖尿病、甲状腺功能减退、无效造血以及骨髓增生、甲状旁腺功能障碍、铁对成骨细胞的毒性作用、生长激素(GH)和胰岛素样生长因子-1(IGF-1)缺乏等因素所致。这些因素通过干扰各种分子机制破坏成骨细胞和破骨细胞之间的平衡,导致骨密度降低。鉴于地中海贫血患者骨质疏松症和骨量减少的高患病率以及其发病过程的复杂性,本综述的目的是评估地中海贫血患者骨质疏松症和骨量减少的分子方面,这可能对治疗有帮助。