Hensman James, Papastamoulis Panagiotis, Glaus Peter, Honkela Antti, Rattray Magnus
Sheffield Institute for Translational Neuroscience (SITraN), Sheffield, UK.
Faculty of Life Sciences.
Bioinformatics. 2015 Dec 15;31(24):3881-9. doi: 10.1093/bioinformatics/btv483. Epub 2015 Aug 26.
Assigning RNA-seq reads to their transcript of origin is a fundamental task in transcript expression estimation. Where ambiguities in assignments exist due to transcripts sharing sequence, e.g. alternative isoforms or alleles, the problem can be solved through probabilistic inference. Bayesian methods have been shown to provide accurate transcript abundance estimates compared with competing methods. However, exact Bayesian inference is intractable and approximate methods such as Markov chain Monte Carlo and Variational Bayes (VB) are typically used. While providing a high degree of accuracy and modelling flexibility, standard implementations can be prohibitively slow for large datasets and complex transcriptome annotations.
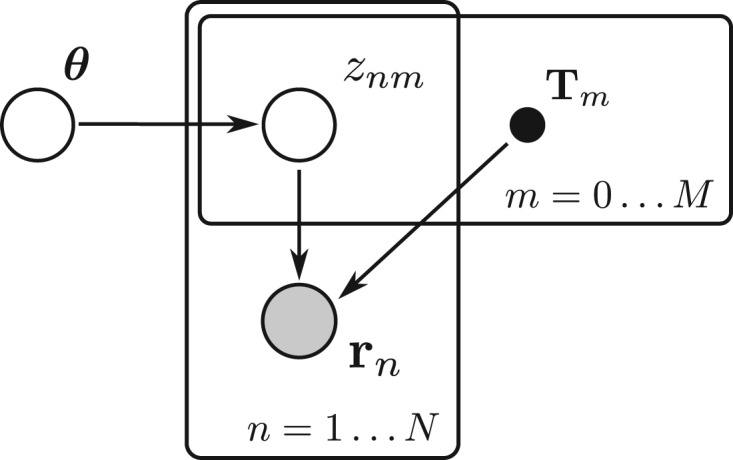
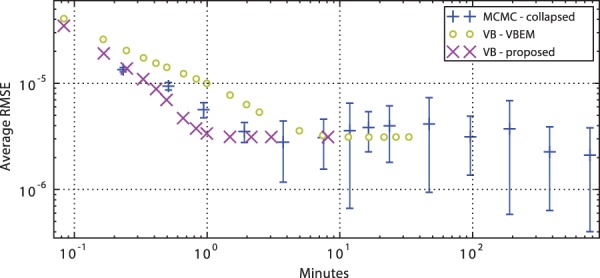
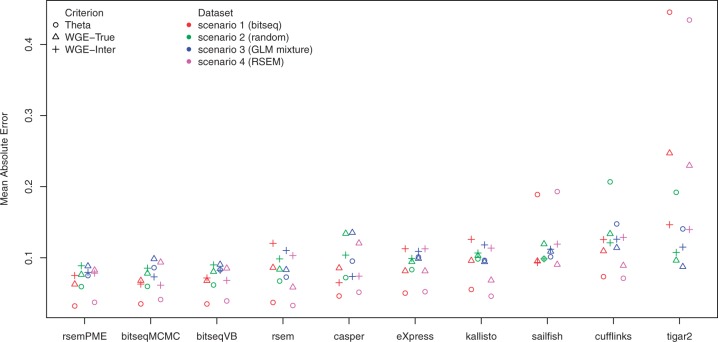
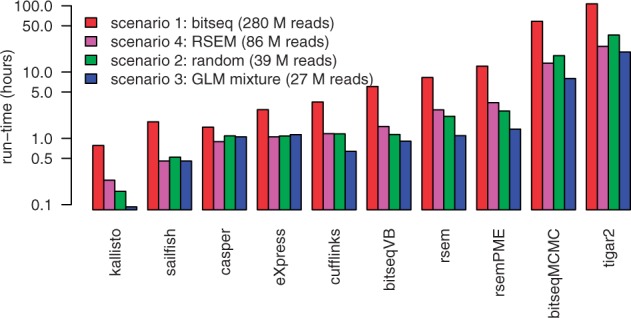
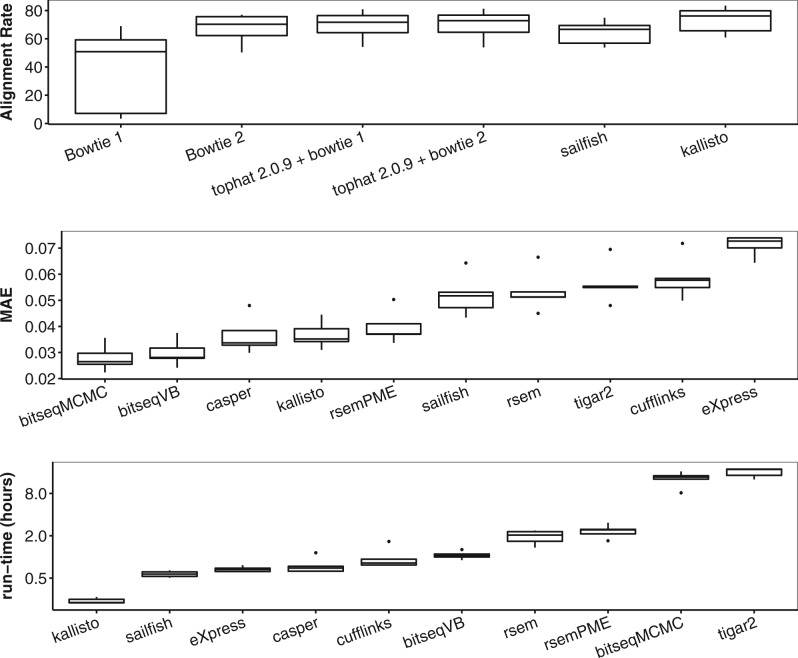
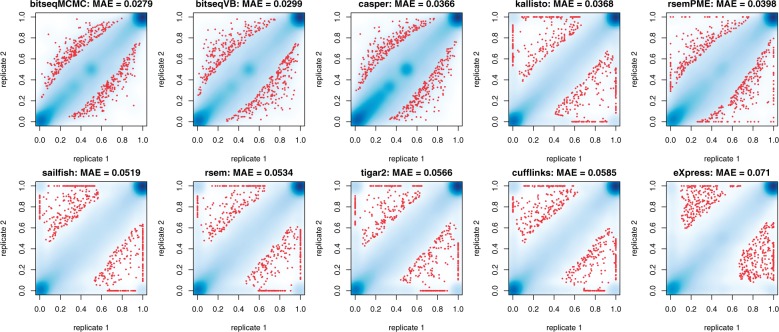
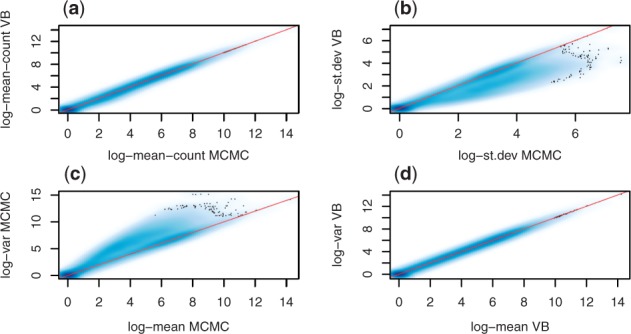
We propose a novel approximate inference scheme based on VB and apply it to an existing model of transcript expression inference from RNA-seq data. Recent advances in VB algorithmics are used to improve the convergence of the algorithm beyond the standard Variational Bayes Expectation Maximization algorithm. We apply our algorithm to simulated and biological datasets, demonstrating a significant increase in speed with only very small loss in accuracy of expression level estimation. We carry out a comparative study against seven popular alternative methods and demonstrate that our new algorithm provides excellent accuracy and inter-replicate consistency while remaining competitive in computation time.
The methods were implemented in R and C++, and are available as part of the BitSeq project at github.com/BitSeq. The method is also available through the BitSeq Bioconductor package. The source code to reproduce all simulation results can be accessed via github.com/BitSeq/BitSeqVB_benchmarking.
将RNA测序读数与其起源转录本进行匹配是转录本表达估计中的一项基本任务。当由于转录本共享序列(例如可变剪接异构体或等位基因)而存在分配模糊性时,可以通过概率推理来解决该问题。与其他竞争方法相比,贝叶斯方法已被证明能提供准确的转录本丰度估计。然而,精确的贝叶斯推理是难以处理的,通常使用马尔可夫链蒙特卡罗和变分贝叶斯(VB)等近似方法。虽然这些方法具有高度的准确性和建模灵活性,但对于大型数据集和复杂的转录组注释,标准实现可能会非常缓慢。
我们提出了一种基于VB的新型近似推理方案,并将其应用于从RNA测序数据进行转录本表达推理的现有模型。VB算法的最新进展被用于改进算法的收敛性,超越了标准的变分贝叶斯期望最大化算法。我们将我们的算法应用于模拟和生物数据集,结果表明在表达水平估计的准确性仅有非常小的损失的情况下,速度有显著提高。我们与七种流行的替代方法进行了比较研究,结果表明我们的新算法在计算时间上具有竞争力的同时,还提供了出色的准确性和重复间的一致性。
这些方法是用R和C++实现的,可作为github.com/BitSeq上BitSeq项目的一部分获取。该方法也可以通过BitSeq Bioconductor包获得。可通过github.com/BitSeq/BitSeqVB_benchmarking访问用于重现所有模拟结果的源代码。