Nariai Naoki, Kojima Kaname, Mimori Takahiro, Sato Yukuto, Kawai Yosuke, Yamaguchi-Kabata Yumi, Nagasaki Masao
BMC Genomics. 2014;15 Suppl 10(Suppl 10):S5. doi: 10.1186/1471-2164-15-S10-S5. Epub 2014 Dec 12.
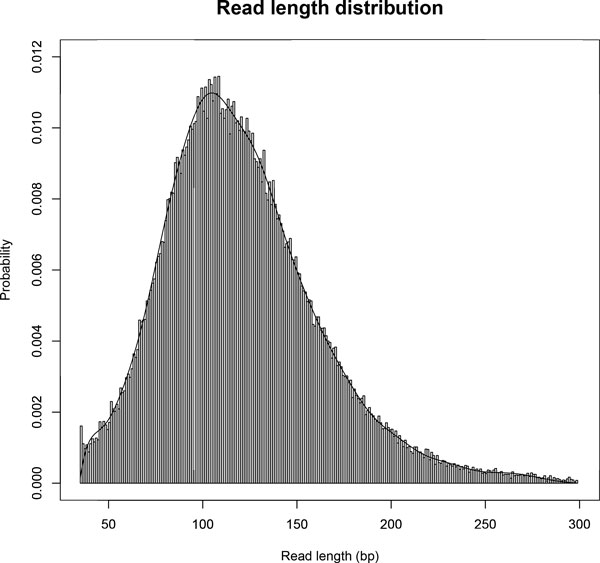
High-throughput RNA sequencing (RNA-Seq) enables quantification and identification of transcripts at single-base resolution. Recently, longer sequence reads become available thanks to the development of new types of sequencing technologies as well as improvements in chemical reagents for the Next Generation Sequencers. Although several computational methods have been proposed for quantifying gene expression levels from RNA-Seq data, they are not sufficiently optimized for longer reads (e.g. >250 bp).
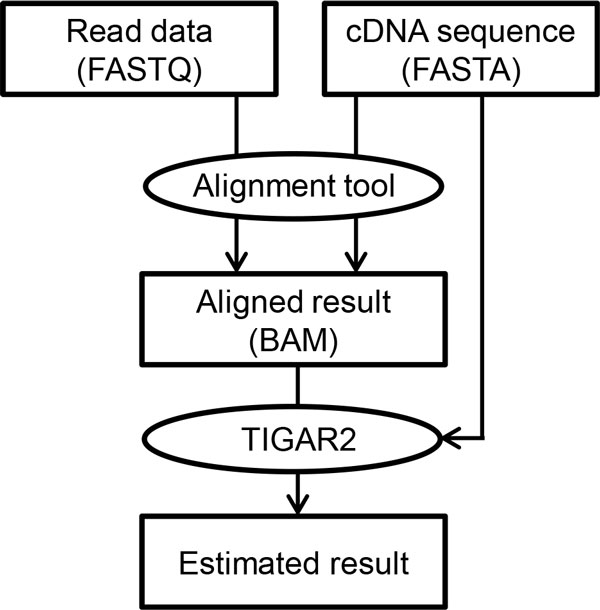
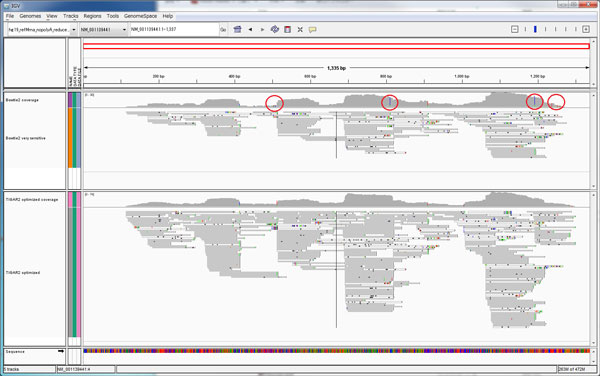
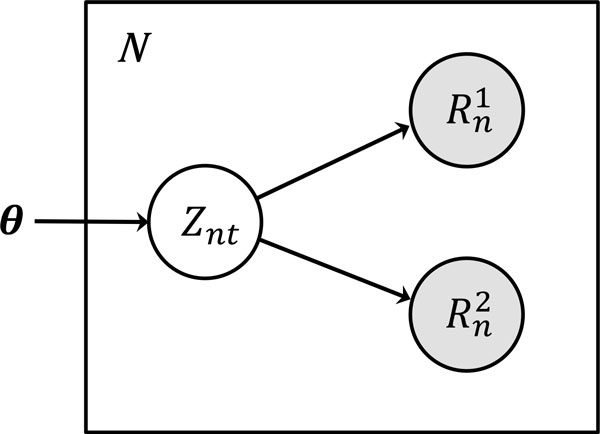
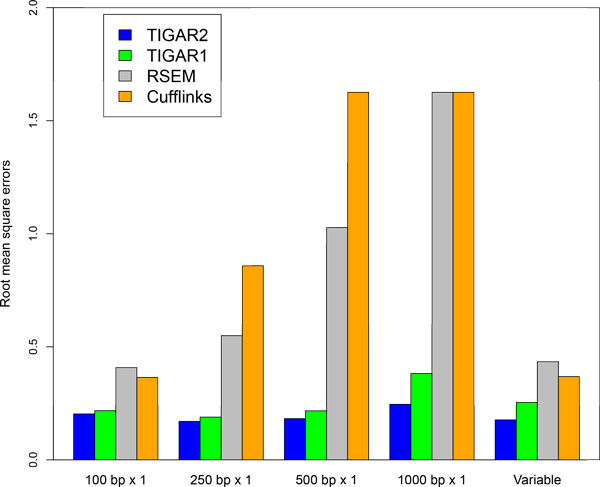
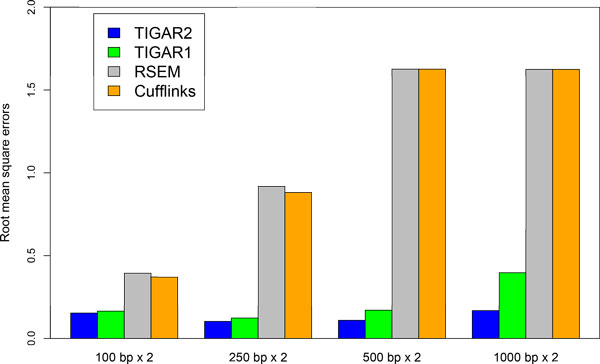
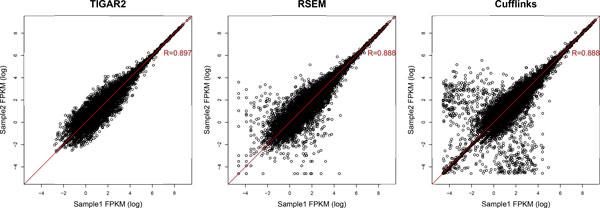
We propose TIGAR2, a statistical method for quantifying transcript isoforms from fixed and variable length RNA-Seq data. Our method models substitution, deletion, and insertion errors of sequencers based on gapped-alignments of reads to the reference cDNA sequences so that sensitive read-aligners such as Bowtie2 and BWA-MEM are effectively incorporated in our pipeline. Also, a heuristic algorithm is implemented in variational Bayesian inference for faster computation. We apply TIGAR2 to both simulation data and real data of human samples and evaluate performance of transcript quantification with TIGAR2 in comparison to existing methods.
TIGAR2 is a sensitive and accurate tool for quantifying transcript isoform abundances from RNA-Seq data. Our method performs better than existing methods for the fixed-length reads (100 bp, 250 bp, 500 bp, and 1000 bp of both single-end and paired-end) and variable-length reads, especially for reads longer than 250 bp.
高通量RNA测序(RNA-Seq)能够在单碱基分辨率下对转录本进行定量和鉴定。近来,由于新型测序技术的发展以及新一代测序仪化学试剂的改进,可获得更长的序列 reads。尽管已经提出了几种从RNA-Seq数据中定量基因表达水平的计算方法,但它们对于更长的reads(例如>250 bp)并未进行充分优化。
我们提出了TIGAR2,一种用于从固定长度和可变长度RNA-Seq数据中定量转录本异构体的统计方法。我们的方法基于reads与参考cDNA序列的间隙比对,对测序仪的替换、缺失和插入错误进行建模,以便将诸如Bowtie2和BWA-MEM等灵敏的read-aligner有效地纳入我们的流程。此外,在变分贝叶斯推理中实现了一种启发式算法以加快计算速度。我们将TIGAR2应用于人类样本的模拟数据和真实数据,并与现有方法相比评估TIGAR2在转录本定量方面的性能。
TIGAR2是一种用于从RNA-Seq数据中定量转录本异构体丰度的灵敏且准确的工具。我们的方法在固定长度reads(单端和双端的100 bp、250 bp、500 bp和1000 bp)和可变长度reads方面比现有方法表现更好,尤其是对于长度超过250 bp的reads。