Li Yafang, Rao Xiayu, Mattox William W, Amos Christopher I, Liu Bin
Department of Biomedical Data Science, Geisel School of Medicine, Dartmouth College, Hanover, New Hampshire, 03755, United States of America.
Center for Genetics and Genomics, Department of Genetics, The University of Texas MD Anderson Cancer Center, Houston, Texas, 77030, United States of America.
PLoS One. 2015 Sep 1;10(9):e0136653. doi: 10.1371/journal.pone.0136653. eCollection 2015.
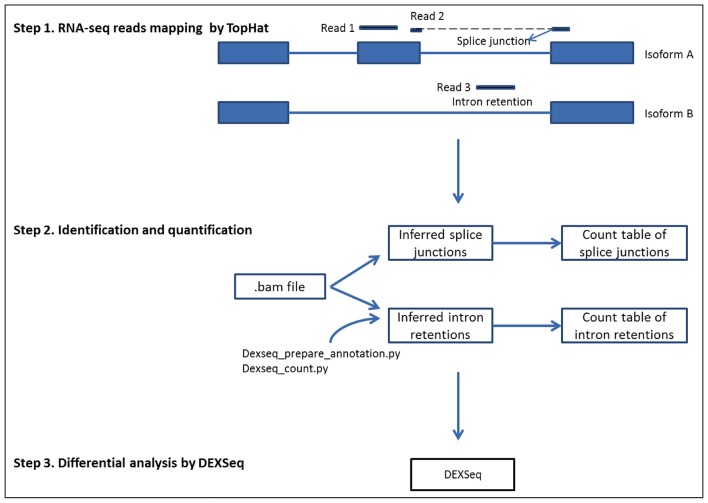
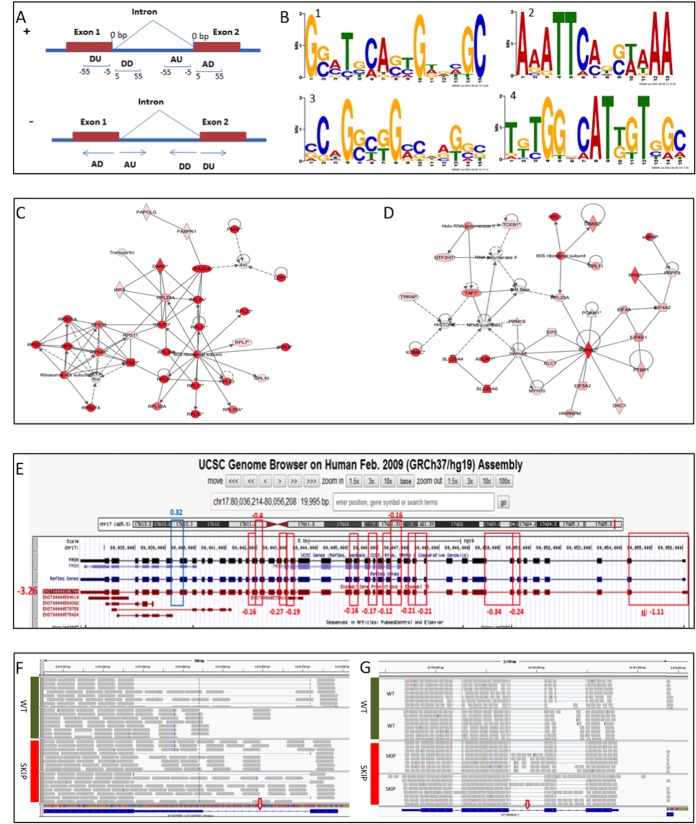
Alternative splicing is an important biological process in the generation of multiple functional transcripts from the same genomic sequences. Differential analysis of splice junctions (SJs) and intron retentions (IRs) is helpful in the detection of alternative splicing events. In this study, we conducted differential analysis of SJs and IRs by use of DEXSeq, a Bioconductor package originally designed for differential exon usage analysis in RNA-seq data analysis. We set up an analysis pipeline including mapping of RNA-seq reads, the preparation of count tables of SJs and IRs as the input files, and the differential analysis in DEXSeq. We analyzed the public RNA-seq datasets generated from RNAi experiments on Drosophila melanogaster S2-DRSC cells to deplete RNA-binding proteins (GSE18508). The analysis confirmed previous findings on the alternative splicing of the trol and Ant2 (sesB) genes in the CG8144 (ps)-depletion experiment and identified some new alternative splicing events in other RNAi experiments. We also identified IRs that were confirmed in our SJ analysis. The proposed method used in our study can output the genomic coordinates of differentially used SJs and thus enable sequence motif search. Sequence motif search and gene function annotation analysis helped us infer the underlying mechanism in alternative splicing events. To further evaluate this method, we also applied the method to public RNA-seq data from human breast cancer (GSE45419) and the plant Arabidopsis (SRP008262). In conclusion, our study showed that DEXSeq can be adapted to differential analysis of SJs and IRs, which will facilitate the identification of alternative splicing events and provide insights into the molecular mechanisms of transcription processes and disease development.
可变剪接是一个重要的生物学过程,可从相同的基因组序列产生多个功能转录本。对剪接位点(SJ)和内含子保留(IR)进行差异分析,有助于检测可变剪接事件。在本研究中,我们使用DEXSeq进行SJ和IR的差异分析,DEXSeq是一个最初设计用于RNA测序数据分析中差异外显子使用分析的Bioconductor软件包。我们建立了一个分析流程,包括RNA测序读数的映射、SJ和IR计数表的制备作为输入文件,以及在DEXSeq中进行差异分析。我们分析了从果蝇S2-DRSC细胞的RNA干扰实验中产生的公共RNA测序数据集,以耗尽RNA结合蛋白(GSE18508)。该分析证实了之前在CG8144(ps)耗尽实验中关于trol和Ant2(sesB)基因可变剪接的发现,并在其他RNA干扰实验中鉴定了一些新可变剪接事件。我们还鉴定了在SJ分析中得到证实的IR。我们研究中使用的方法可以输出差异使用的SJ的基因组坐标,从而实现序列基序搜索。序列基序搜索和基因功能注释分析帮助我们推断可变剪接事件的潜在机制。为了进一步评估该方法,我们还将该方法应用于来自人类乳腺癌(GSE45419)和植物拟南芥(SRP008262)的公共RNA测序数据。总之,我们的研究表明DEXSeq可适用于SJ和IR的差异分析,这将有助于识别可变剪接事件,并为转录过程和疾病发展的分子机制提供见解。