Hayer Katharina E, Pizarro Angel, Lahens Nicholas F, Hogenesch John B, Grant Gregory R
University of Pennsylvania, Institute for Translational Medicine and Therapeutics, Philadelphia, PA 19104.
Scientific Computing at Amazon Web Services, Seattle, WA 98108.
Bioinformatics. 2015 Dec 15;31(24):3938-45. doi: 10.1093/bioinformatics/btv488. Epub 2015 Sep 3.
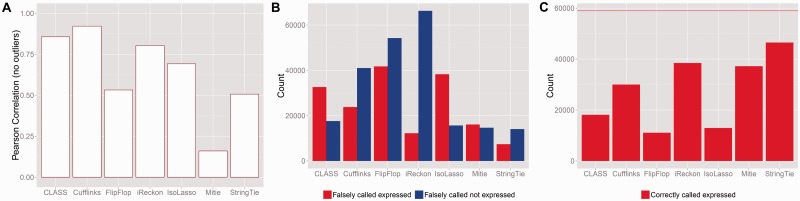
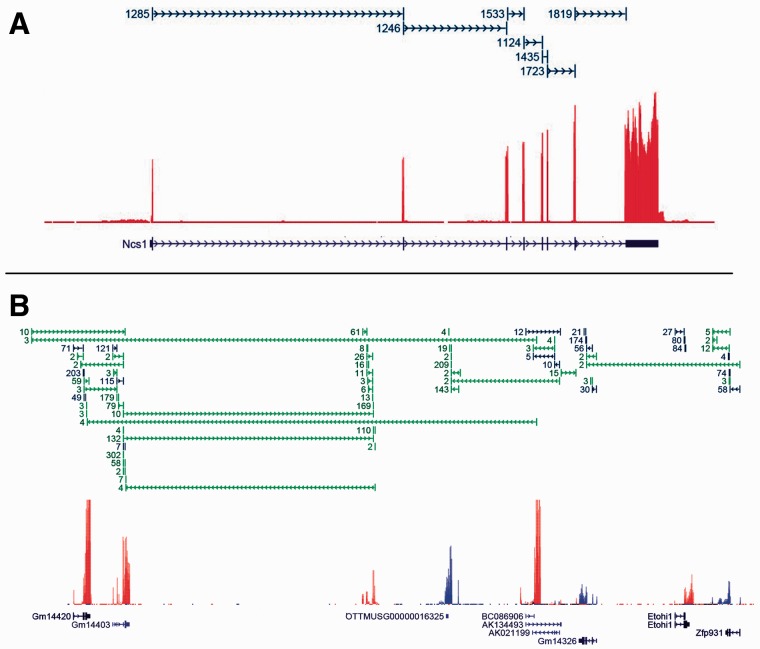
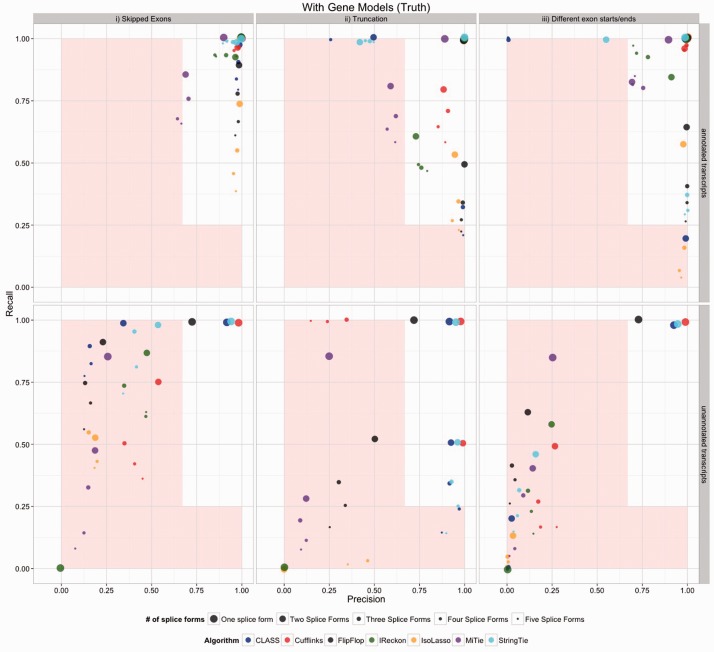
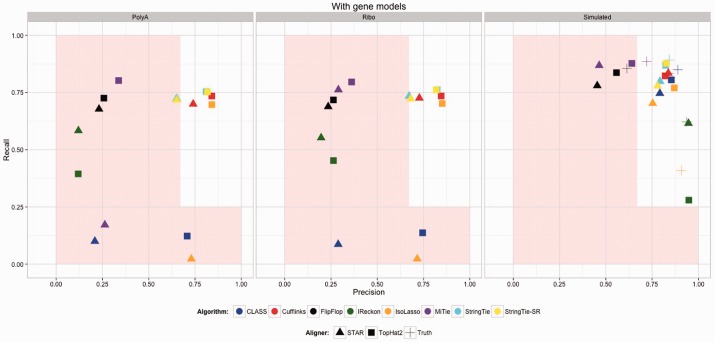
Because of the advantages of RNA sequencing (RNA-Seq) over microarrays, it is gaining widespread popularity for highly parallel gene expression analysis. For example, RNA-Seq is expected to be able to provide accurate identification and quantification of full-length splice forms. A number of informatics packages have been developed for this purpose, but short reads make it a difficult problem in principle. Sequencing error and polymorphisms add further complications. It has become necessary to perform studies to determine which algorithms perform best and which if any algorithms perform adequately. However, there is a dearth of independent and unbiased benchmarking studies. Here we take an approach using both simulated and experimental benchmark data to evaluate their accuracy.
We conclude that most methods are inaccurate even using idealized data, and that no method is highly accurate once multiple splice forms, polymorphisms, intron signal, sequencing errors, alignment errors, annotation errors and other complicating factors are present. These results point to the pressing need for further algorithm development.
Simulated datasets and other supporting information can be found at http://bioinf.itmat.upenn.edu/BEERS/bp2.
Supplementary data are available at Bioinformatics online.
由于RNA测序(RNA-Seq)相对于微阵列的优势,它在高度并行的基因表达分析中越来越受欢迎。例如,RNA-Seq有望能够准确识别和定量全长剪接形式。为此已经开发了许多信息学软件包,但短读长在原则上使其成为一个难题。测序错误和多态性增加了进一步的复杂性。有必要进行研究以确定哪些算法表现最佳,以及是否有任何算法表现足够好。然而,缺乏独立且无偏见的基准研究。在这里,我们采用一种使用模拟和实验基准数据的方法来评估它们的准确性。
我们得出结论,即使使用理想化数据,大多数方法也不准确,并且一旦存在多种剪接形式、多态性、内含子信号、测序错误、比对错误、注释错误和其他复杂因素,就没有方法是高度准确的。这些结果表明迫切需要进一步开发算法。
模拟数据集和其他支持信息可在http://bioinf.itmat.upenn.edu/BEERS/bp2找到。
补充数据可在《生物信息学》在线获取。