Galardini Marco, Brilli Matteo, Spini Giulia, Rossi Matteo, Roncaglia Bianca, Bani Alessia, Chiancianesi Manuela, Moretto Marco, Engelen Kristof, Bacci Giovanni, Pini Francesco, Biondi Emanuele G, Bazzicalupo Marco, Mengoni Alessio
Department of Biology, University of Florence, Florence, Italy.
Department of Genomics and Biology of Fruit Crops, Research and Innovation Centre, Fondazione Edmund Mach (FEM), San Michele all'Adige, Italy.
PLoS Comput Biol. 2015 Sep 4;11(9):e1004478. doi: 10.1371/journal.pcbi.1004478. eCollection 2015 Sep.
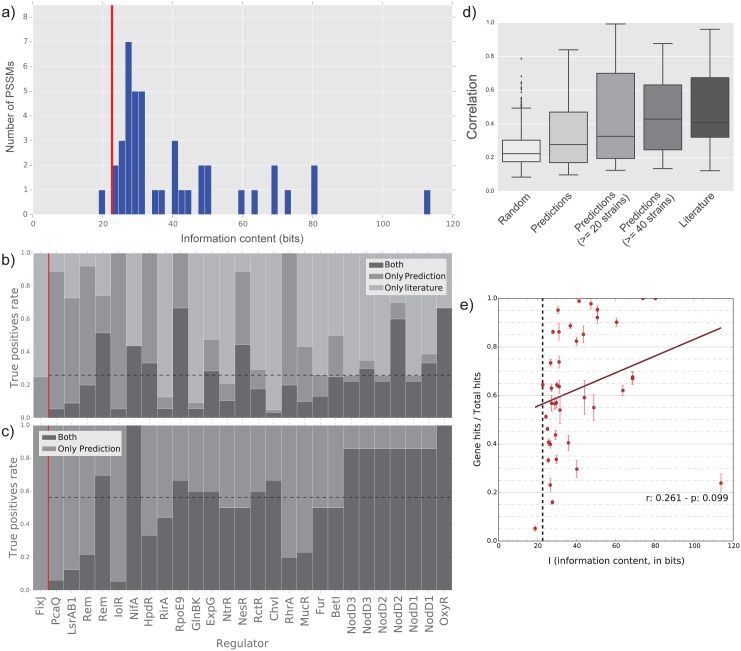
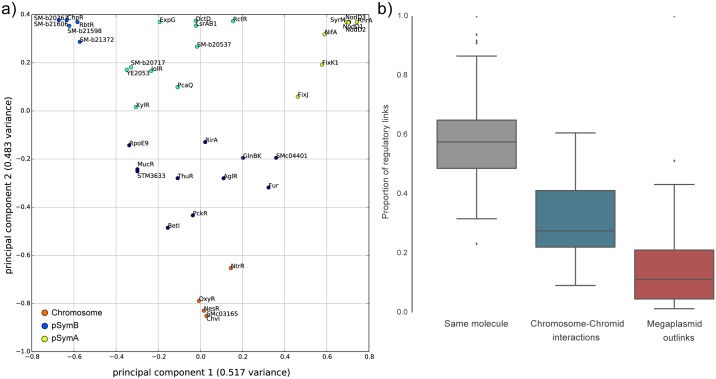
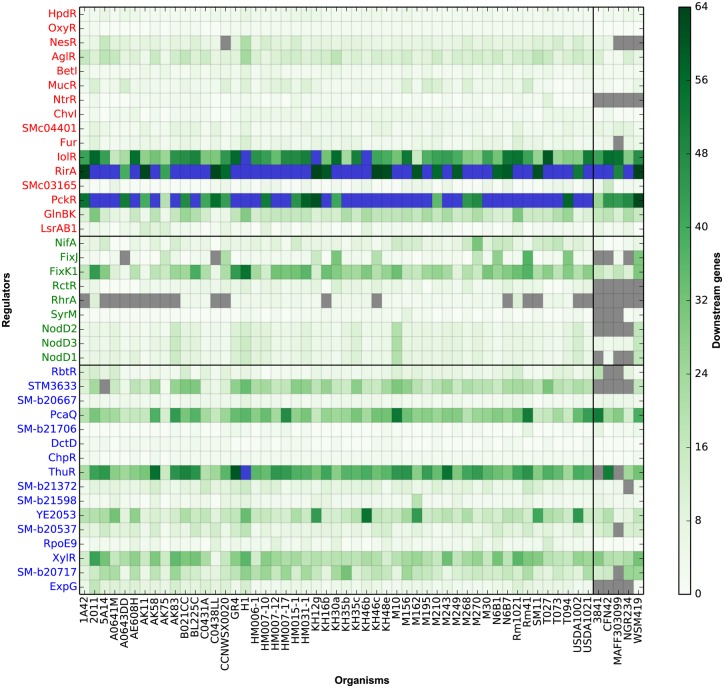
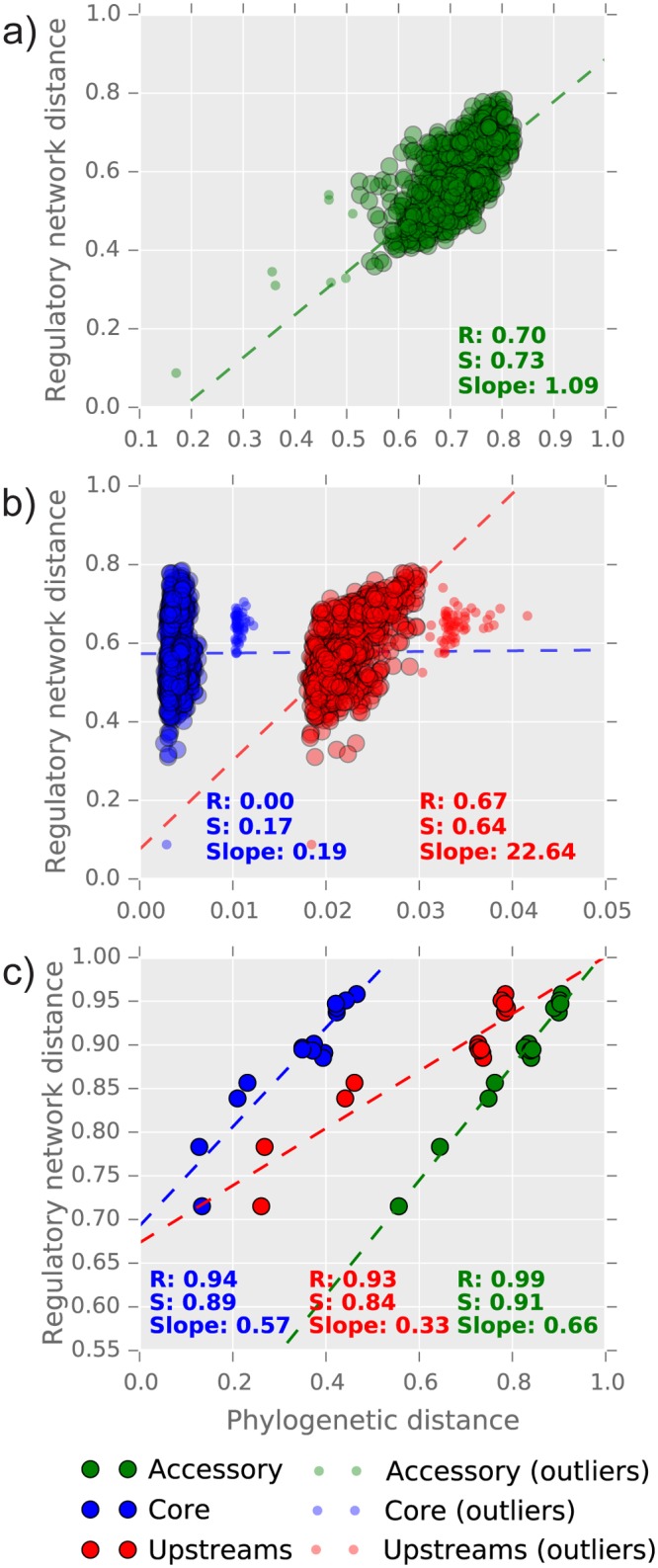
Reconstruction of the regulatory network is an important step in understanding how organisms control the expression of gene products and therefore phenotypes. Recent studies have pointed out the importance of regulatory network plasticity in bacterial adaptation and evolution. The evolution of such networks within and outside the species boundary is however still obscure. Sinorhizobium meliloti is an ideal species for such study, having three large replicons, many genomes available and a significant knowledge of its transcription factors (TF). Each replicon has a specific functional and evolutionary mark; which might also emerge from the analysis of their regulatory signatures. Here we have studied the plasticity of the regulatory network within and outside the S. meliloti species, looking for the presence of 41 TFs binding motifs in 51 strains and 5 related rhizobial species. We have detected a preference of several TFs for one of the three replicons, and the function of regulated genes was found to be in accordance with the overall replicon functional signature: house-keeping functions for the chromosome, metabolism for the chromid, symbiosis for the megaplasmid. This therefore suggests a replicon-specific wiring of the regulatory network in the S. meliloti species. At the same time a significant part of the predicted regulatory network is shared between the chromosome and the chromid, thus adding an additional layer by which the chromid integrates itself in the core genome. Furthermore, the regulatory network distance was found to be correlated with both promoter regions and accessory genome evolution inside the species, indicating that both pangenome compartments are involved in the regulatory network evolution. We also observed that genes which are not included in the species regulatory network are more likely to belong to the accessory genome, indicating that regulatory interactions should also be considered to predict gene conservation in bacterial pangenomes.
重建调控网络是理解生物体如何控制基因产物表达进而控制表型的重要一步。最近的研究指出了调控网络可塑性在细菌适应和进化中的重要性。然而,这种网络在物种边界内外的进化仍然不清楚。苜蓿中华根瘤菌是进行此类研究的理想物种,它有三个大的复制子,有许多可用的基因组,并且对其转录因子(TF)有相当多的了解。每个复制子都有特定的功能和进化标记;这也可能从它们的调控特征分析中显现出来。在这里,我们研究了苜蓿中华根瘤菌物种内外调控网络的可塑性,在51个菌株和5个相关根瘤菌物种中寻找41个转录因子结合基序的存在情况。我们检测到几个转录因子对三个复制子之一有偏好,并且发现受调控基因的功能与整体复制子功能特征一致:染色体负责管家功能,质粒负责代谢,大质粒负责共生。因此,这表明苜蓿中华根瘤菌物种的调控网络存在复制子特异性的连接方式。同时,预测的调控网络的很大一部分在染色体和质粒之间共享,从而增加了一层质粒将自身整合到核心基因组中的方式。此外,发现调控网络距离与物种内部的启动子区域和辅助基因组进化都相关,表明两个泛基因组部分都参与了调控网络进化。我们还观察到,不包含在物种调控网络中的基因更有可能属于辅助基因组,这表明在预测细菌泛基因组中的基因保守性时也应考虑调控相互作用。