Galán-Vásquez Edgardo, Perez-Rueda Ernesto
Departamento de Ingeniería de Sistemas Computacionales y Automatización, Instituto de Investigaciones en Matemáticas Aplicadas y en Sistemas, Ciudad Universitaria, Universidad Nacional Autónoma de México, Ciudad de México, Mexico.
Instituto de Investigaciones en Matemáticas Aplicadas y en Sistemas, Universidad Nacional Autónoma de México, Unidad Académica Yucatán, Mérida, Mexico.
Front Mol Biosci. 2019 Dec 13;6:139. doi: 10.3389/fmolb.2019.00139. eCollection 2019.
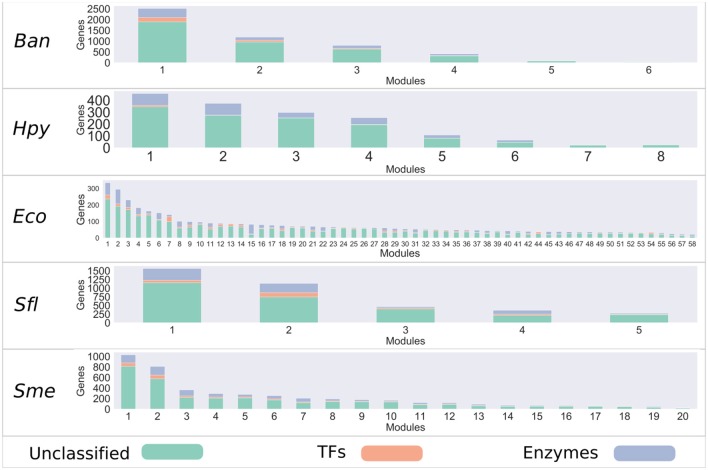
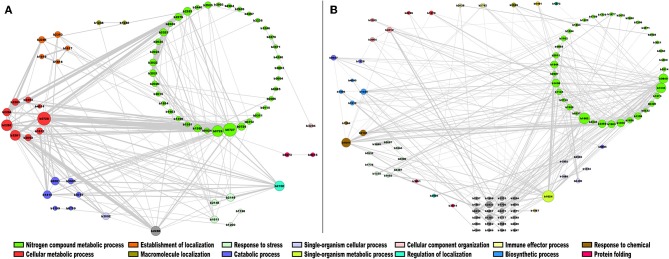
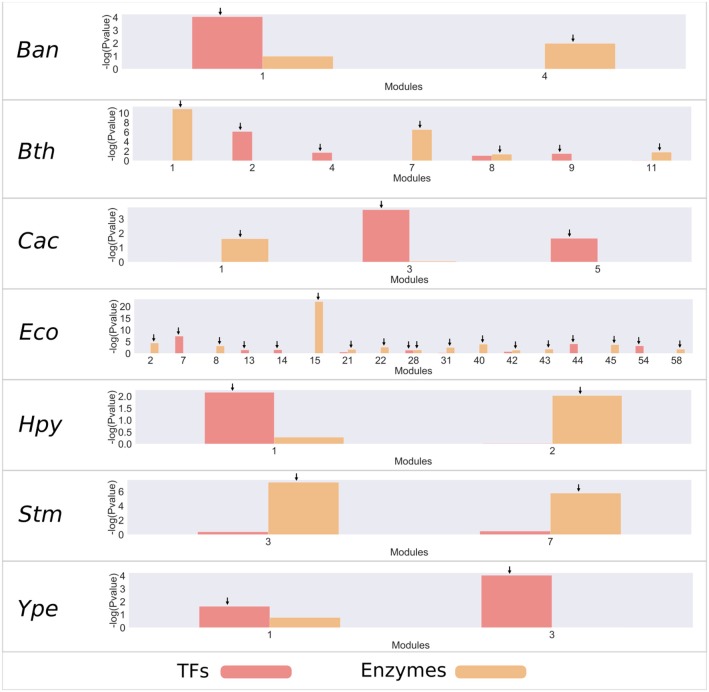
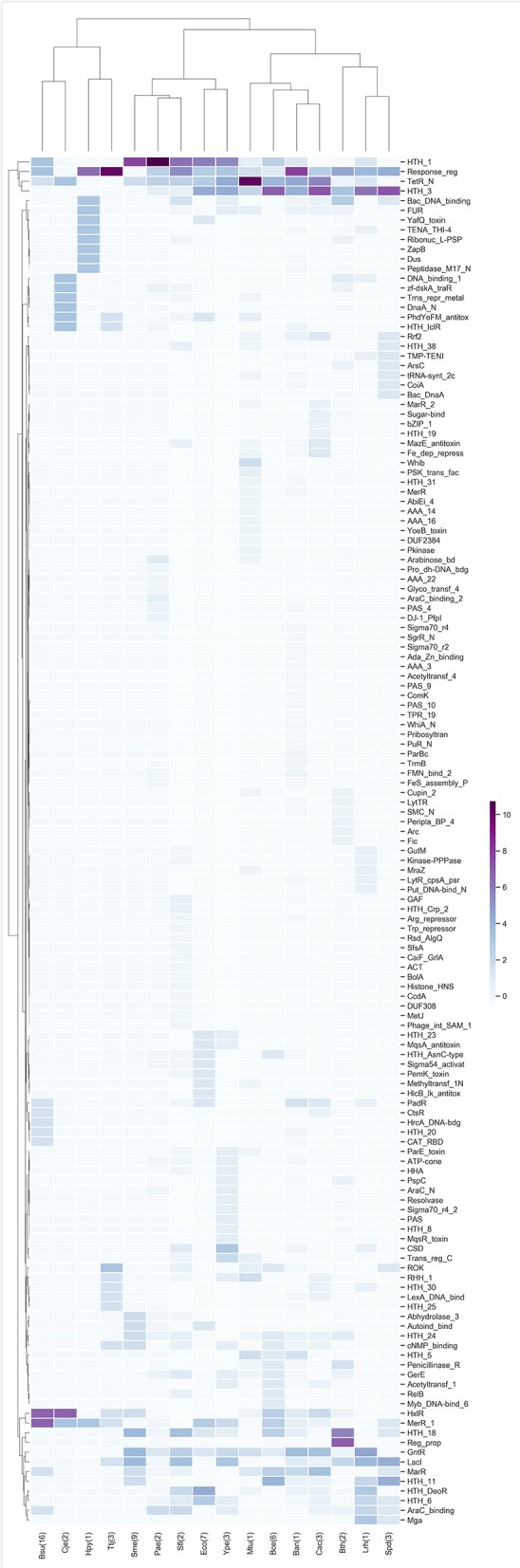
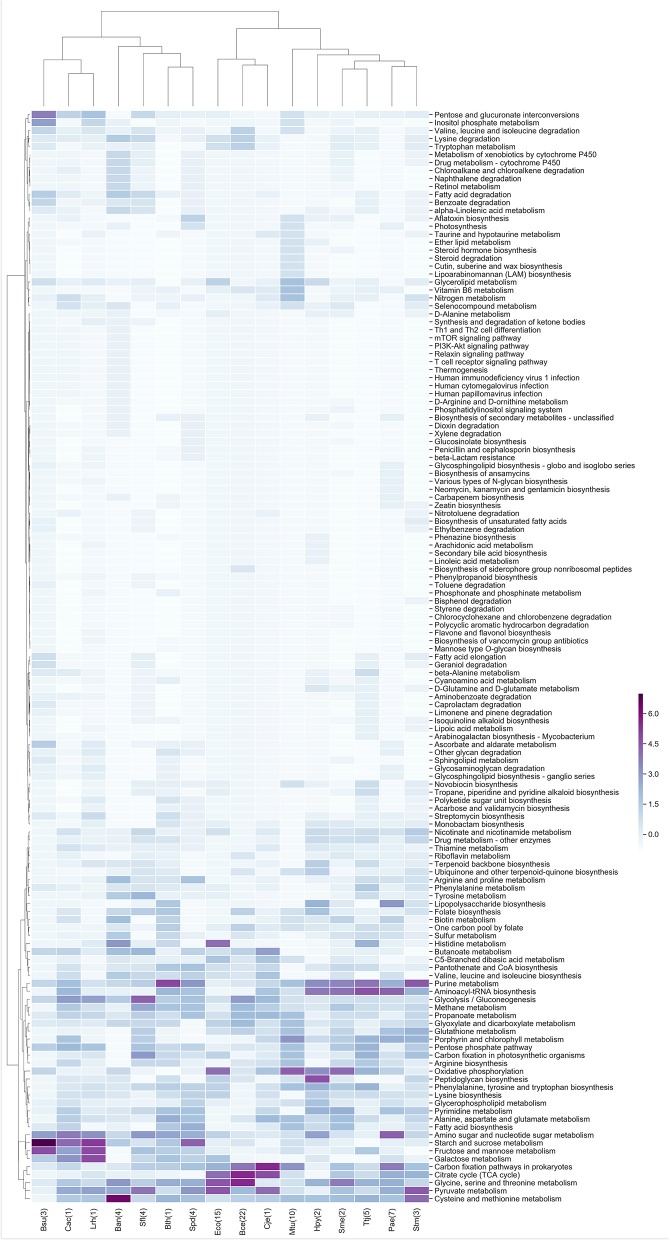
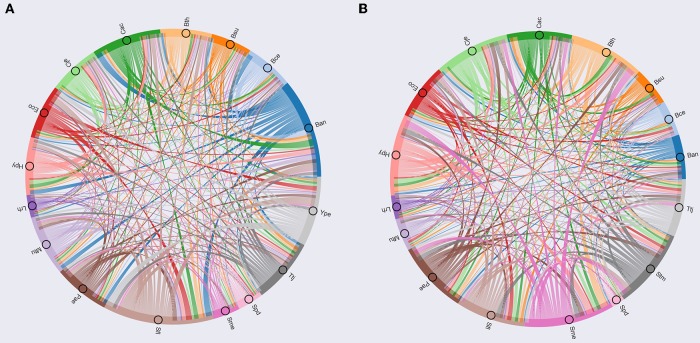
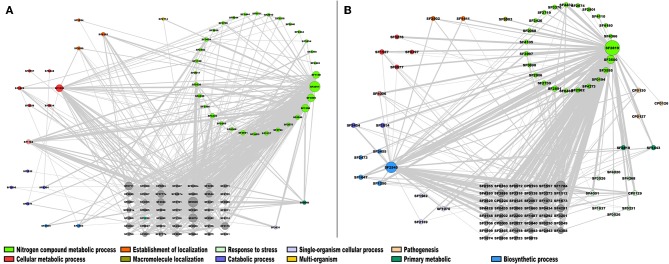
Biological systems respond to environmental perturbations and to a large diversity of compounds through gene interactions, and these genetic factors comprise complex networks. In particular, a wide variety of gene co-expression networks have been constructed in recent years thanks to the dramatic increase of experimental information obtained with techniques, such as microarrays and RNA sequencing. These networks allow the identification of groups of co-expressed genes that can function in the same process and, in turn, these networks may be related to biological functions of industrial, medical and academic interest. In this study, gene co-expression networks for 17 bacterial organisms from the COLOMBOS database were analyzed via weighted gene co-expression network analysis and clustered into modules of genes with similar expression patterns for each species. These networks were analyzed to determine relevant modules through a hypergeometric approach based on a set of transcription factors and enzymes for each genome. The richest modules were characterized using PFAM families and KEGG metabolic maps. Additionally, we conducted a Gene Ontology analysis for enrichment of biological functions. Finally, we identified modules that shared similarity through all the studied organisms by using comparative genomics.
生物系统通过基因相互作用对环境扰动和多种多样的化合物作出反应,并且这些遗传因素构成复杂的网络。特别是,近年来由于诸如微阵列和RNA测序等技术所获得的实验信息急剧增加,已经构建了各种各样的基因共表达网络。这些网络能够识别可在同一过程中发挥作用的共表达基因群体,进而,这些网络可能与工业、医学和学术领域感兴趣的生物学功能相关。在本研究中,通过加权基因共表达网络分析对来自COLOMBOS数据库的17种细菌生物体的基因共表达网络进行了分析,并针对每个物种聚集成具有相似表达模式的基因模块。基于每个基因组的一组转录因子和酶,通过超几何方法对这些网络进行分析以确定相关模块。使用PFAM家族和KEGG代谢图谱对最丰富的模块进行了表征。此外,我们进行了基因本体分析以富集生物学功能。最后,我们通过比较基因组学鉴定了所有研究生物体中具有相似性的模块。