From the Institute of Clinical Biochemistry and Pathobiochemistry, University Hospital Würzburg (S.M.C., M.K., C.C., A.Z.), Rudolf Virchow Center (H.D.M., M.B., H.M.H.), Institute of Virology and Immunobiology (M.B.L.), and Division of Hepatology, Medical Clinic II, University Hospital Würzburg (H.M.H.), University of Würzburg, Würzburg, Germany; Department of Pathology, Maastricht University Medical Centre, Cardiovascular Research Institute Maastricht, Maastricht, The Netherlands (J.C.S., T.L.T.); Joslin Diabetes Center, Harvard Medical School, Boston, MA (C.C.-F., S.K.); Experimental and Molecular Pediatric Cardiology, German Heart Center Munich, TU Munich, Munich, Germany (F.V., A.G.); Department of Vascular and Endovascular Surgery, Klinikum Rechts der Isar der Technischen Universität München, Munich, Germany (J.P.); and Department of Pathology, Amsterdam Medical Centre, Amsterdam, The Netherlands (M.J.D.).
Arterioscler Thromb Vasc Biol. 2015 Nov;35(11):2316-25. doi: 10.1161/ATVBAHA.115.306171. Epub 2015 Sep 24.
Although immune responses drive the pathogenesis of atherosclerosis, mechanisms that control antigen-presenting cell (APC)-mediated immune activation in atherosclerosis remain elusive. We here investigated the function of hypoxia-inducible factor (HIF)-1α in APCs in atherosclerosis.
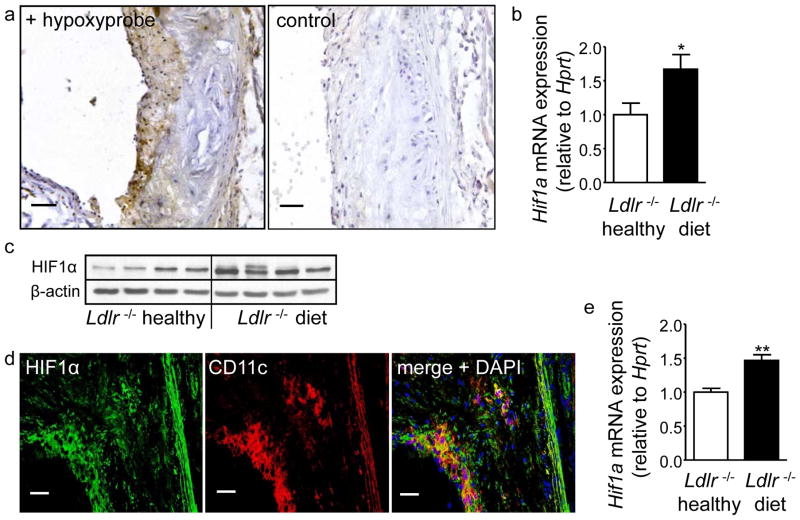
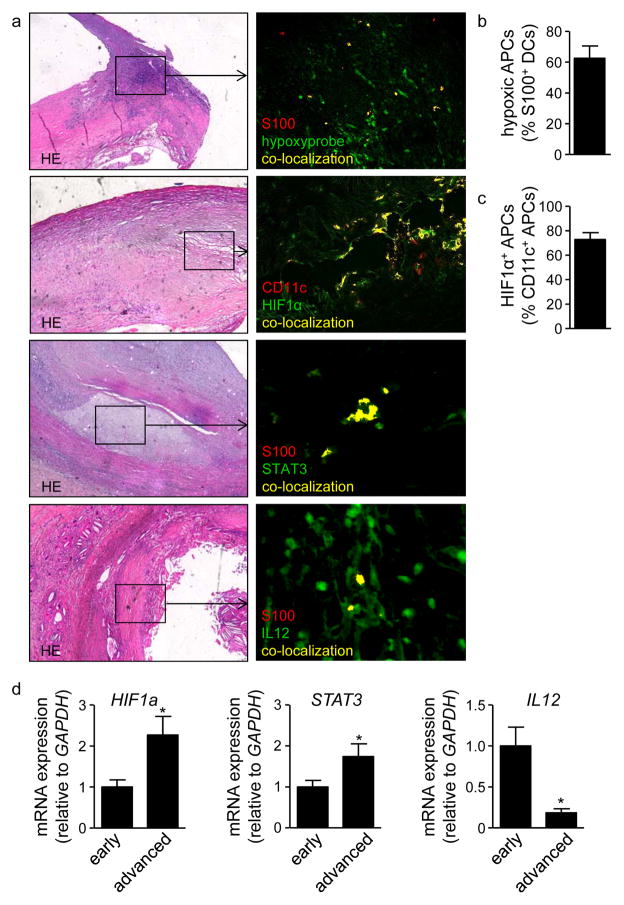
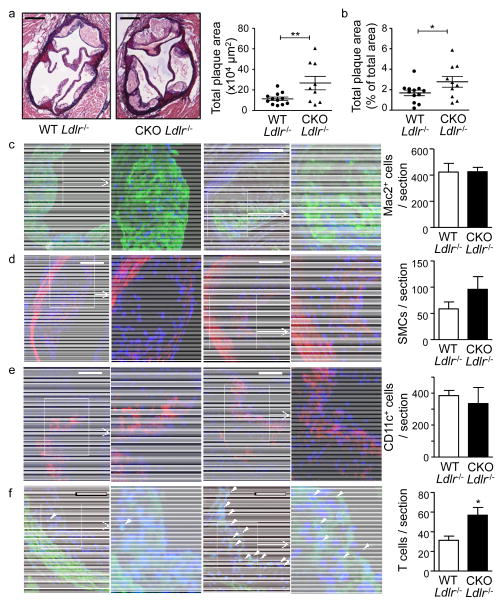
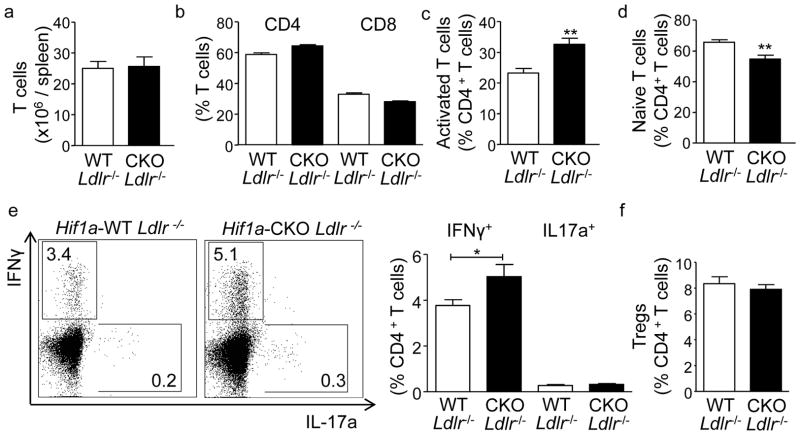
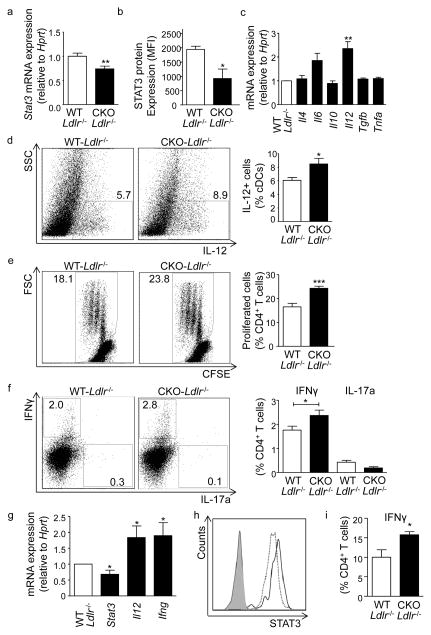
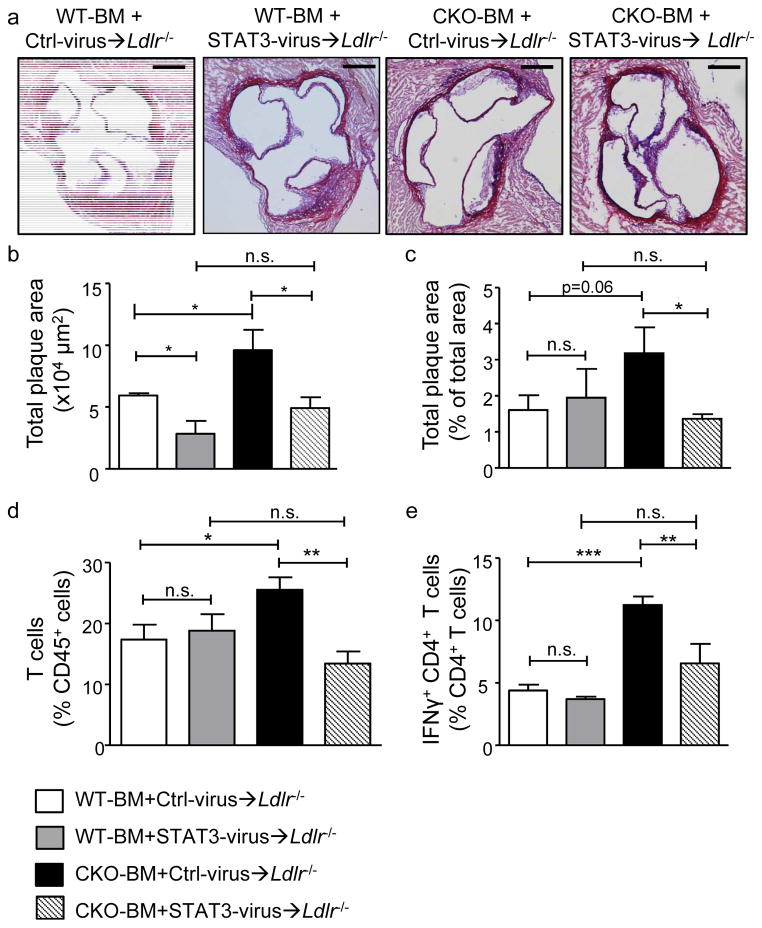
We found upregulated HIF1α expression in CD11c(+) APCs within atherosclerotic plaques of low-density lipoprotein receptor-deficient (Ldlr(-/-)) mice. Conditional deletion of Hif1a in CD11c(+) APCs in high-fat diet-fed Ldlr(-/-) mice accelerated atherosclerotic plaque formation and increased lesional T-cell infiltrates, revealing a protective role of this transcription factor. HIF1α directly controls Signal Transducers and Activators of Transcription 3 (Stat3), and a reduced STAT3 expression was found in HIF1α-deficient APCs and aortic tissue, together with an upregulated interleukin-12 expression and expansion of type 1 T-helper (Th1) cells. Overexpression of STAT3 in Hif1a-deficient APCs in bone marrow reversed enhanced atherosclerotic lesion formation and reduced Th1 cell expansion in chimeric Ldlr(-/-) mice. Notably, deletion of Hif1a in LysM(+) bone marrow cells in Ldlr(-/-) mice did not affect lesion formation or T-cell activation. In human atherosclerotic lesions, HIF1α, STAT3, and interleukin-12 protein were found to colocalize with APCs.
Our findings identify HIF1α to antagonize APC activation and Th1 T cell polarization during atherogenesis in Ldlr(-/-) mice and to attenuate the progression of atherosclerosis. These data substantiate the critical role of APCs in controlling immune mechanisms that drive atherosclerotic lesion development.
尽管免疫反应驱动动脉粥样硬化的发病机制,但控制抗原呈递细胞(APC)介导的动脉粥样硬化中免疫激活的机制仍不清楚。我们在此研究了缺氧诱导因子(HIF)-1α在动脉粥样硬化中的 APC 中的功能。
我们发现,在低密度脂蛋白受体缺陷(Ldlr(-/-))小鼠的动脉粥样硬化斑块中,CD11c(+)APC 中 HIF1α表达上调。高脂饮食喂养的 Ldlr(-/-)小鼠中 CD11c(+)APC 中 Hif1a 的条件缺失加速了动脉粥样硬化斑块的形成,并增加了病变中的 T 细胞浸润,表明该转录因子具有保护作用。HIF1α直接控制信号转导子和转录激活子 3(Stat3),并且在 HIF1α缺陷 APC 和主动脉组织中发现 Stat3 表达降低,同时伴有白细胞介素-12 表达上调和 1 型 T 辅助(Th1)细胞扩增。在骨髓中 Hif1a 缺陷 APC 中转染 STAT3 可逆转嵌合 Ldlr(-/-)小鼠中增强的动脉粥样硬化病变形成和减少 Th1 细胞扩增。值得注意的是,在 Ldlr(-/-)小鼠的 LysM(+)骨髓细胞中缺失 Hif1a 并不影响病变形成或 T 细胞激活。在人类动脉粥样硬化病变中,发现 HIF1α、STAT3 和白细胞介素-12 蛋白与 APC 共定位。
我们的发现确定 HIF1α拮抗 Ldlr(-/-)小鼠动脉粥样硬化过程中 APC 的激活和 Th1 T 细胞极化,并减弱动脉粥样硬化的进展。这些数据证实了 APC 在控制驱动动脉粥样硬化病变发展的免疫机制中的关键作用。