Asimit Jennifer L, Panoutsopoulou Kalliope, Wheeler Eleanor, Berndt Sonja I, Cordell Heather J, Morris Andrew P, Zeggini Eleftheria, Barroso Inês
Wellcome Trust Sanger Institute, Hinxton, Cambridge, United Kingdom.
Division of Cancer Epidemiology and Genetics, National Cancer Institute, US National Institutes of Health, Bethesda, Maryland, United States of America.
Genet Epidemiol. 2015 Dec;39(8):624-34. doi: 10.1002/gepi.21919. Epub 2015 Sep 28.
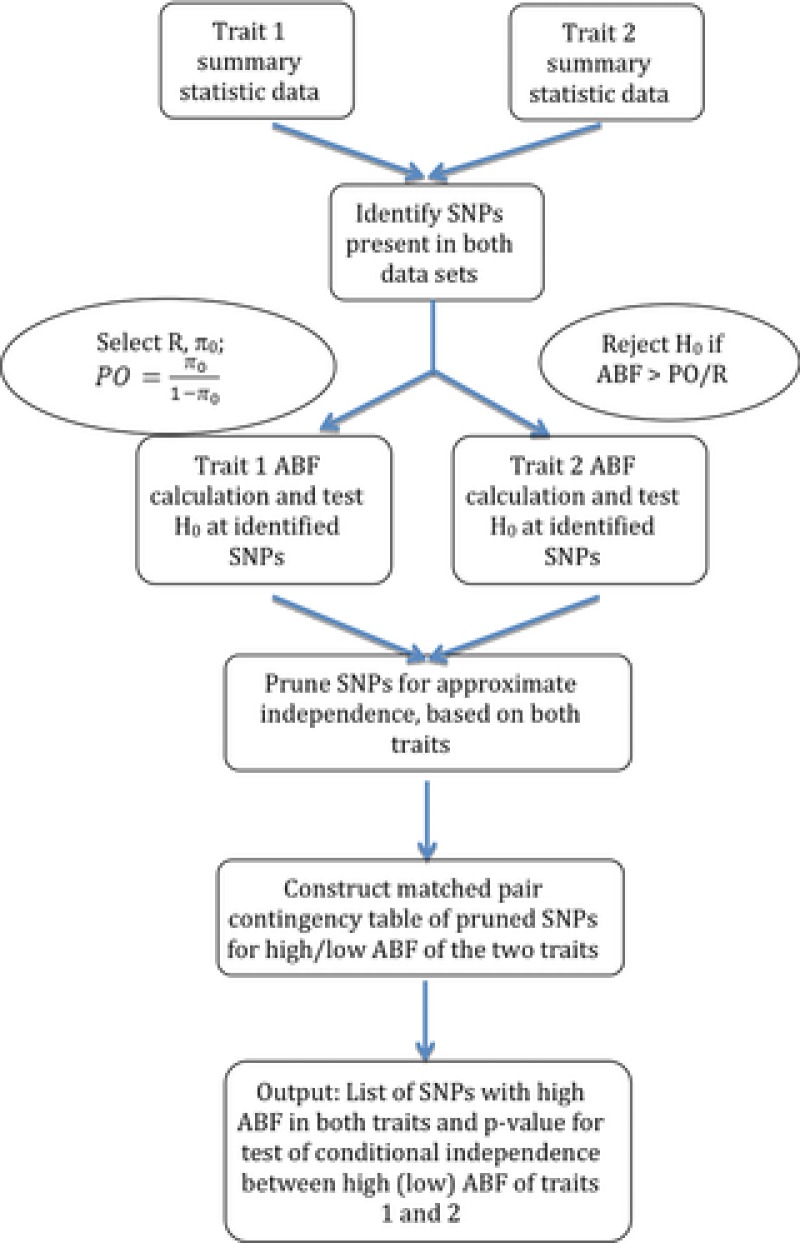
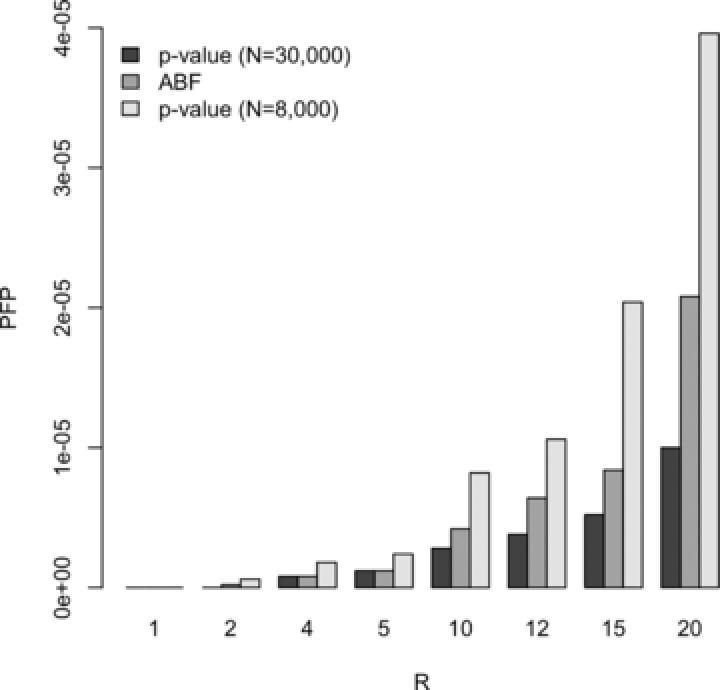
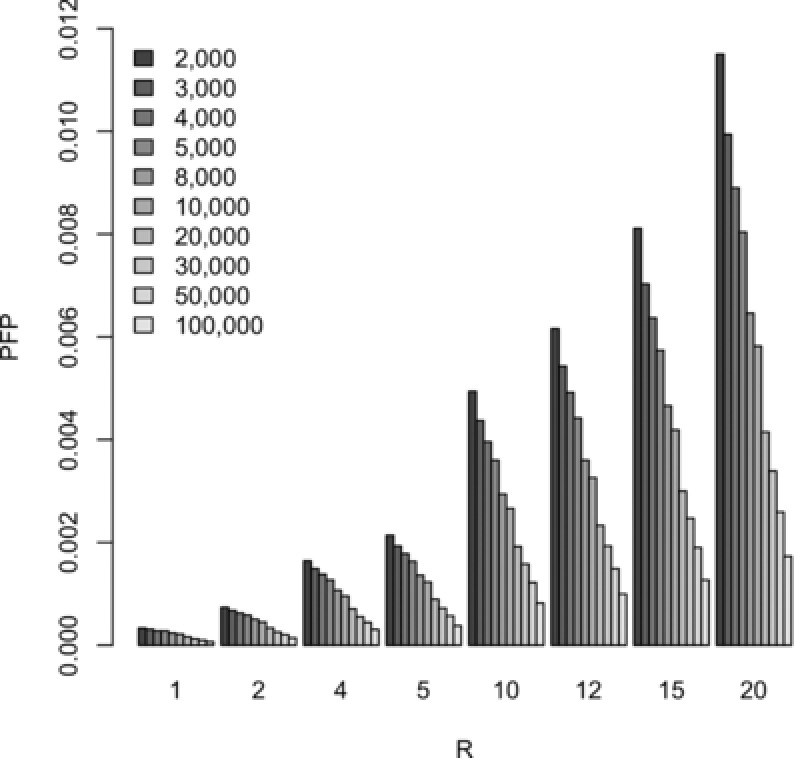
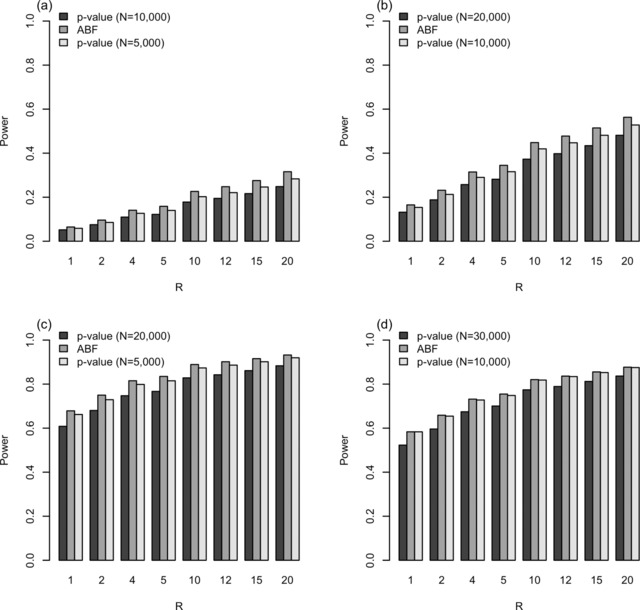
Diseases often cooccur in individuals more often than expected by chance, and may be explained by shared underlying genetic etiology. A common approach to genetic overlap analyses is to use summary genome-wide association study data to identify single-nucleotide polymorphisms (SNPs) that are associated with multiple traits at a selected P-value threshold. However, P-values do not account for differences in power, whereas Bayes' factors (BFs) do, and may be approximated using summary statistics. We use simulation studies to compare the power of frequentist and Bayesian approaches with overlap analyses, and to decide on appropriate thresholds for comparison between the two methods. It is empirically illustrated that BFs have the advantage over P-values of a decreasing type I error rate as study size increases for single-disease associations. Consequently, the overlap analysis of traits from different-sized studies encounters issues in fair P-value threshold selection, whereas BFs are adjusted automatically. Extensive simulations show that Bayesian overlap analyses tend to have higher power than those that assess association strength with P-values, particularly in low-power scenarios. Calibration tables between BFs and P-values are provided for a range of sample sizes, as well as an approximation approach for sample sizes that are not in the calibration table. Although P-values are sometimes thought more intuitive, these tables assist in removing the opaqueness of Bayesian thresholds and may also be used in the selection of a BF threshold to meet a certain type I error rate. An application of our methods is used to identify variants associated with both obesity and osteoarthritis.
疾病在个体中同时出现的频率往往高于偶然预期,这可能由共同的潜在遗传病因来解释。基因重叠分析的一种常见方法是使用全基因组关联研究的汇总数据,以识别在选定的P值阈值下与多种性状相关的单核苷酸多态性(SNP)。然而,P值没有考虑检验效能的差异,而贝叶斯因子(BF)可以考虑,并且可以使用汇总统计量进行近似计算。我们通过模拟研究来比较频率学派和贝叶斯方法在重叠分析中的效能,并确定两种方法进行比较时的合适阈值。经验表明,对于单病关联,随着研究规模的增加,贝叶斯因子在降低I型错误率方面比P值具有优势。因此,来自不同规模研究的性状重叠分析在公平的P值阈值选择上会遇到问题,而贝叶斯因子会自动调整。大量模拟表明,贝叶斯重叠分析往往比用P值评估关联强度的方法具有更高的效能,尤其是在低效能情况下。我们提供了一系列样本量下贝叶斯因子和P值之间的校准表,以及针对不在校准表中的样本量的近似方法。尽管有时认为P值更直观,但这些表格有助于消除贝叶斯阈值的不透明性,也可用于选择贝叶斯因子阈值以满足特定的I型错误率。我们的方法应用于识别与肥胖症和骨关节炎都相关的变异。