Bennewitz Jörn, Edel Christian, Fries Ruedi, Meuwissen Theo H E, Wellmann Robin
Institute of Animal Science, University of Hohenheim, 70593, Stuttgart, Germany.
Institute of Animal Breeding, Bavarian State Research Center for Agriculture, 85580, Grub, Germany.
Genet Sel Evol. 2017 Jan 14;49(1):7. doi: 10.1186/s12711-017-0284-7.
Multi-marker methods, which fit all markers simultaneously, were originally tailored for genomic selection purposes, but have proven to be useful also in association analyses, especially the so-called BayesC Bayesian methods. In a recent study, BayesD extended BayesC towards accounting for dominance effects and improved prediction accuracy and persistence in genomic selection. The current study investigated the power and precision of BayesC and BayesD in genome-wide association studies by means of stochastic simulations and applied these methods to a dairy cattle dataset.
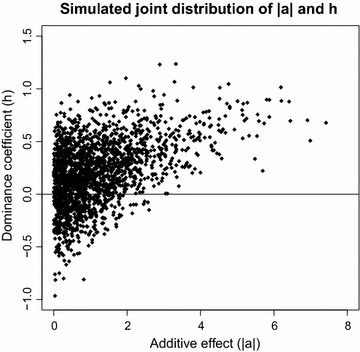
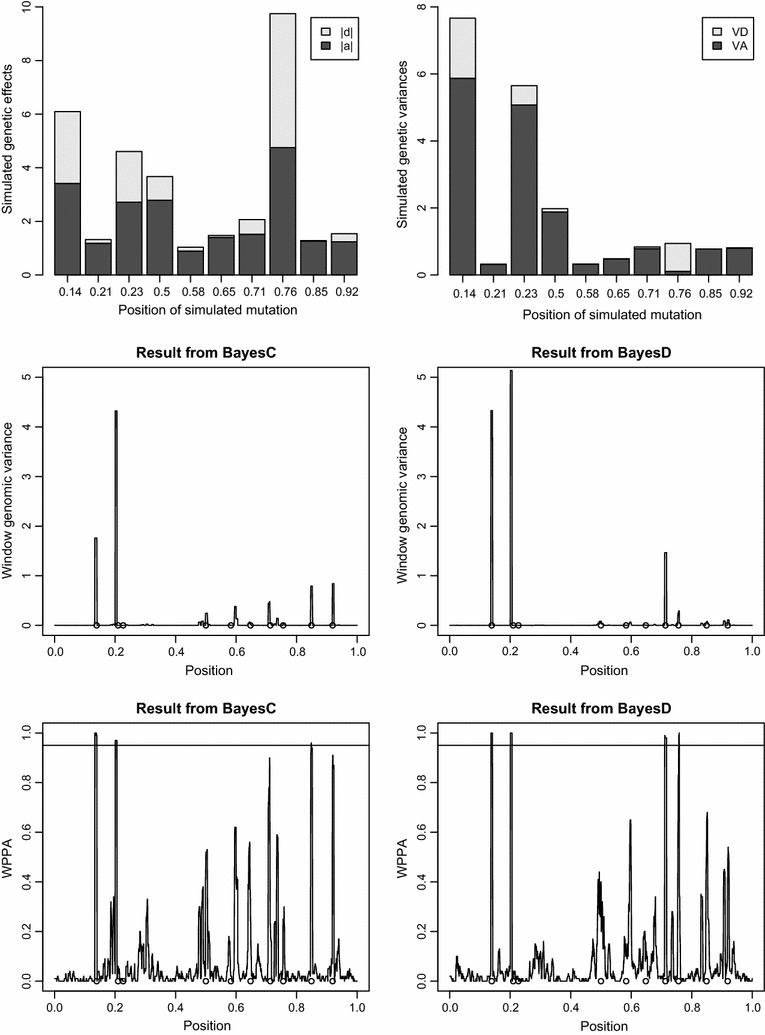
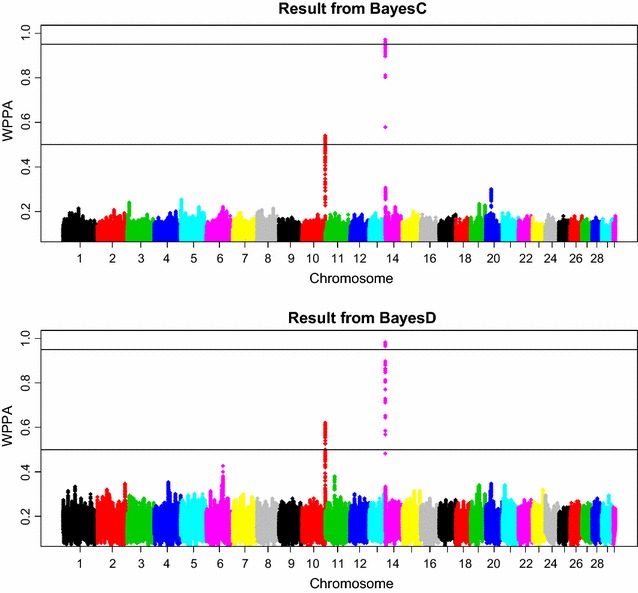
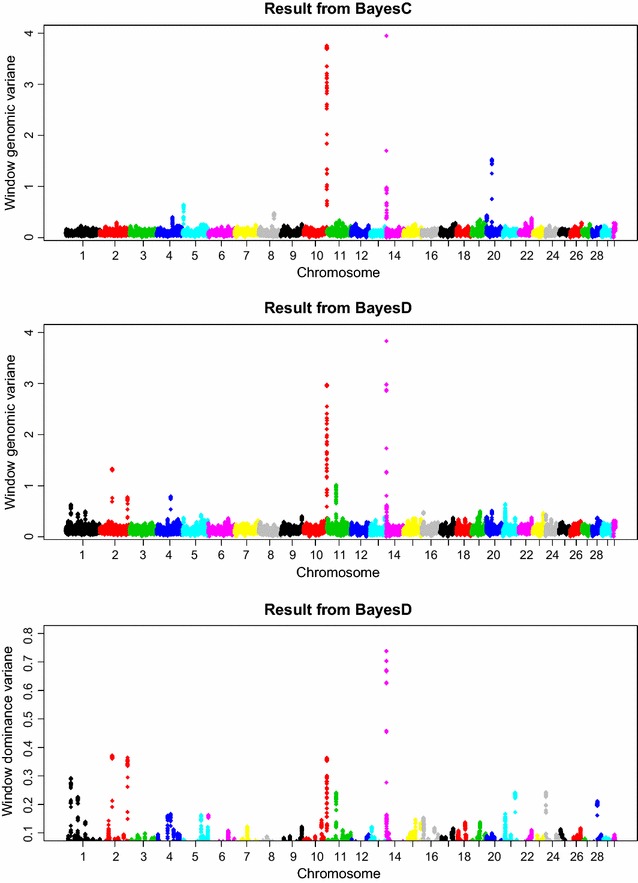
The simulation protocol was designed to mimic the genetic architecture of quantitative traits as realistically as possible. Special emphasis was put on the joint distribution of the additive and dominance effects of causative mutations. Additive marker effects were estimated by BayesC and additive and dominance effects by BayesD. The dependencies between additive and dominance effects were modelled in BayesD by choosing appropriate priors. A sliding-window approach was used. For each window, the R. Fernando window posterior probability of association was calculated and this was used for inference purpose. The power to map segregating causal effects and the mapping precision were assessed for various marker densities up to full sequence information and various window sizes.
Power to map a QTL increased with higher marker densities and larger window sizes. This held true for both methods. Method BayesD had improved power compared to BayesC. The increase in power was between -2 and 8% for causative genes that explained more than 2.5% of the genetic variance. In addition, inspection of the estimates of genomic window dominance variance allowed for inference about the magnitude of dominance at significant associations, which remains hidden in BayesC analysis. Mapping precision was not substantially improved by BayesD.
BayesD improved power, but precision only slightly. Application of BayesD needs large datasets with genotypes and own performance records as phenotypes. Given the current efforts to establish cow reference populations in dairy cattle genomic selection schemes, such datasets are expected to be soon available, which will enable the application of BayesD for association mapping and genomic prediction purposes.
多标记方法可同时拟合所有标记,最初是为基因组选择目的而设计的,但已证明在关联分析中也很有用,特别是所谓的贝叶斯C类贝叶斯方法。在最近的一项研究中,贝叶斯D方法在考虑显性效应方面对贝叶斯C方法进行了扩展,并提高了基因组选择中的预测准确性和持续性。本研究通过随机模拟研究了贝叶斯C和贝叶斯D在全基因组关联研究中的功效和精度,并将这些方法应用于一个奶牛数据集。
模拟方案旨在尽可能逼真地模拟数量性状的遗传结构。特别强调了致病突变的加性效应和显性效应的联合分布。贝叶斯C方法估计加性标记效应,贝叶斯D方法估计加性效应和显性效应。在贝叶斯D方法中,通过选择合适的先验来模拟加性效应和显性效应之间的依赖性。采用滑动窗口方法。对于每个窗口,计算R. Fernando窗口关联后验概率,并将其用于推断目的。评估了高达全序列信息的各种标记密度和各种窗口大小下映射分离因果效应的功效和映射精度。
映射QTL的功效随着标记密度的增加和窗口大小的增大而提高。两种方法都是如此。与贝叶斯C方法相比,贝叶斯D方法的功效有所提高。对于解释超过2.5%遗传方差的致病基因,功效提高了-2%至8%。此外,对基因组窗口显性方差估计值的检查允许推断显著关联处的显性程度,而这在贝叶斯C分析中是隐藏的。贝叶斯D方法并未显著提高映射精度。
贝叶斯D方法提高了功效,但精度仅略有提高。贝叶斯D方法的应用需要具有基因型的大型数据集以及自身的性能记录作为表型。鉴于目前在奶牛基因组选择计划中建立奶牛参考群体的努力,预计很快就能获得此类数据集,这将使贝叶斯D方法能够用于关联图谱绘制和基因组预测目的。