Anjum Samreen, Morganella Sandro, D'Angelo Fulvio, Iavarone Antonio, Ceccarelli Michele
Computational Sciences and Engineering, Qatar Computing Research Institute, Doha, P. O. Box 5825, Qatar.
European Molecular Biology Laboratory, European Bioinformatics Institute, (EMBL -EBI), Wellcome Trust Genome Campus, Cambridge, CB10 1SD, UK.
BMC Bioinformatics. 2015 Sep 29;16:315. doi: 10.1186/s12859-015-0748-0.
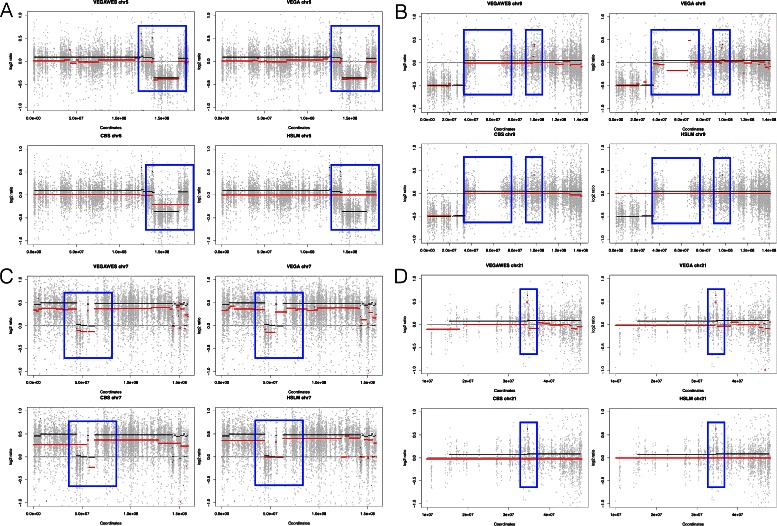
Copy number variations are important in the detection and progression of significant tumors and diseases. Recently, Whole Exome Sequencing is gaining popularity with copy number variations detection due to low cost and better efficiency. In this work, we developed VEGAWES for accurate and robust detection of copy number variations on WES data. VEGAWES is an extension to a variational based segmentation algorithm, VEGA: Variational estimator for genomic aberrations, which has previously outperformed several algorithms on segmenting array comparative genomic hybridization data.
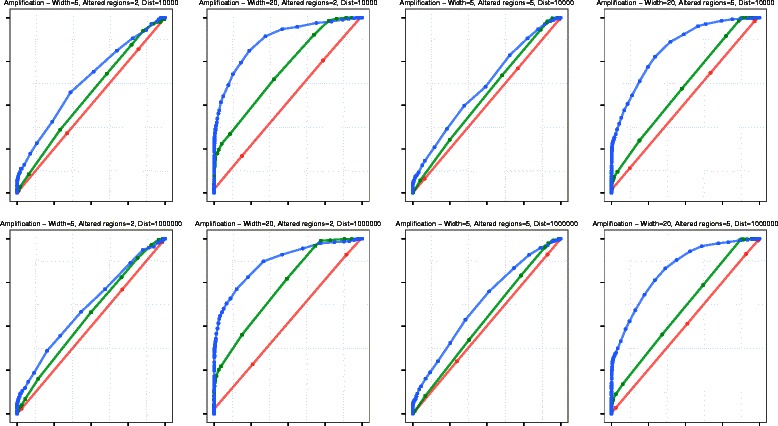
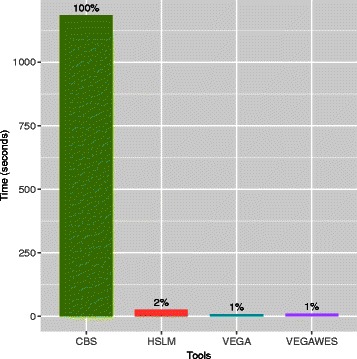
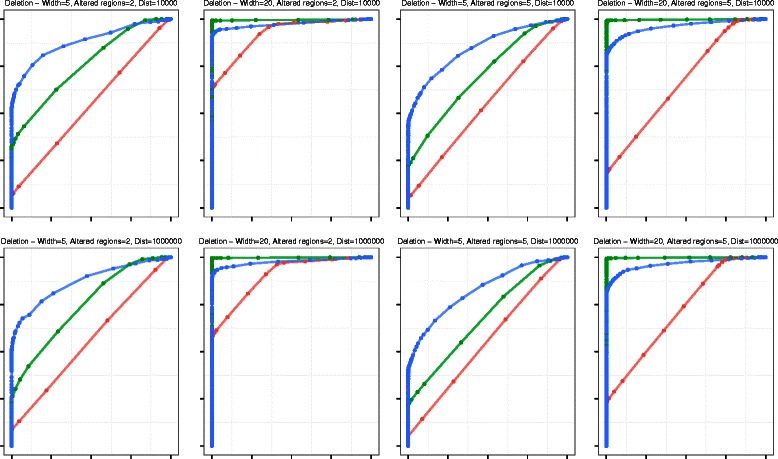
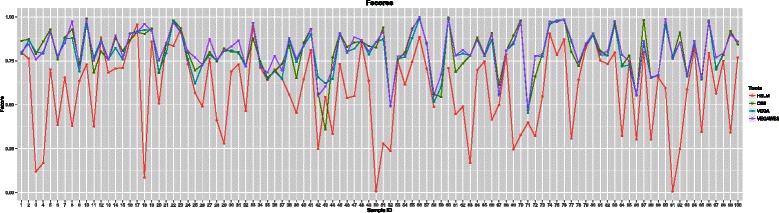
We tested this algorithm on synthetic data and 100 Glioblastoma Multiforme primary tumor samples. The results on the real data were analyzed with segmentation obtained from Single-nucleotide polymorphism data as ground truth. We compared our results with two other segmentation algorithms and assessed the performance based on accuracy and time.
In terms of both accuracy and time, VEGAWES provided better results on the synthetic data and tumor samples demonstrating its potential in robust detection of aberrant regions in the genome.
拷贝数变异在重大肿瘤和疾病的检测及进展中具有重要意义。近来,全外显子组测序因其低成本和更高效率,在拷贝数变异检测方面越来越受欢迎。在本研究中,我们开发了VEGAWES,用于准确且稳健地检测全外显子组测序数据中的拷贝数变异。VEGAWES是基于变分的分割算法VEGA(基因组畸变的变分估计器)的扩展,VEGA此前在分割阵列比较基因组杂交数据方面性能优于其他几种算法。
我们在合成数据和100个多形性胶质母细胞瘤原发肿瘤样本上测试了该算法。以单核苷酸多态性数据获得的分割结果作为真实对照,对真实数据的结果进行分析。我们将自己的结果与其他两种分割算法进行比较,并基于准确性和时间评估性能。
在准确性和时间方面,VEGAWES在合成数据和肿瘤样本上均给出了更好的结果,证明了其在稳健检测基因组异常区域方面的潜力。