Cramaro Wibke J, Revets Dominique, Hunewald Oliver E, Sinner Regina, Reye Anna L, Muller Claude P
Department of Infection and Immunity, Luxembourg Institute of Health (former Centre de Recherche Public de la Santé)/Laboratoire National de Santé, Luxembourg, Luxembourg.
BMC Genomics. 2015 Oct 28;16:871. doi: 10.1186/s12864-015-1981-7.
In Europe, Ixodes ricinus ticks are the most important vectors of diseases threatening humans, livestock, wildlife and companion animals. Nevertheless, genomic sequence information is missing and functional annotation of transcripts and proteins is limited. This lack of information is restricting studies of the vector and its interactions with pathogens and hosts. Here we present and integrate the first analysis of the I. ricinus genome with the transcriptome and proteome of the unfed I. ricinus midgut.
Whole genome sequencing was performed on I. ricinus ticks and the sequences were de novo assembled. In parallel, I. ricinus ticks were dissected and the midgut transcriptome sequenced. Both datasets were integrated by transcript discovery analysis to identify putative genes and genome contigs were screened for homology. An alignment-based and a motif-search-based approach were combined for the annotation of the midgut transcriptome. Additionally, midgut proteins were identified and annotated by mass spectrometry with public databases and the in-house built transcriptome database as references and results were cross-validated.
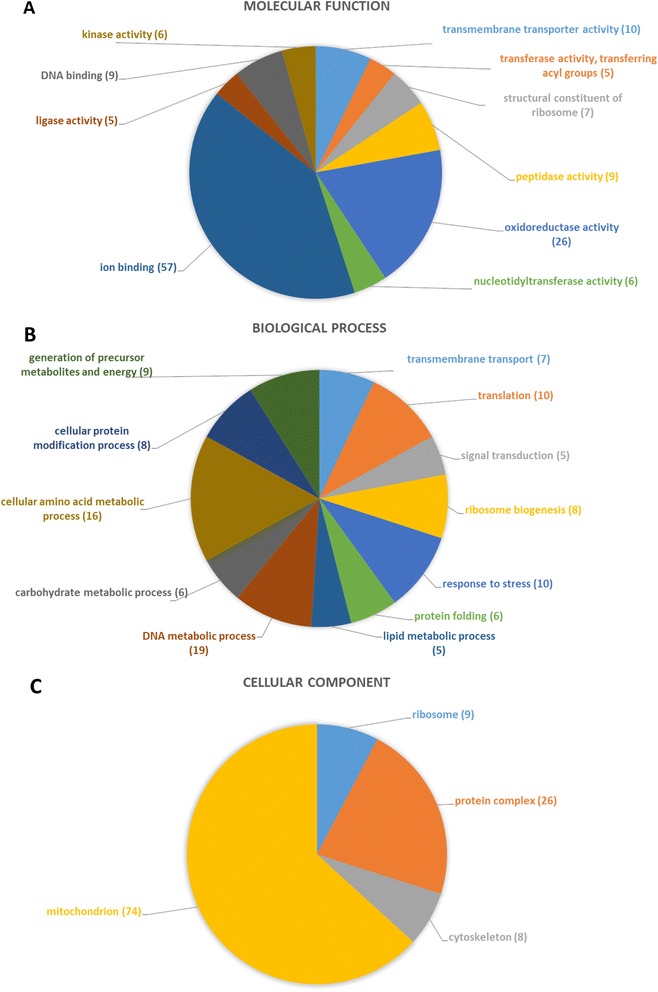
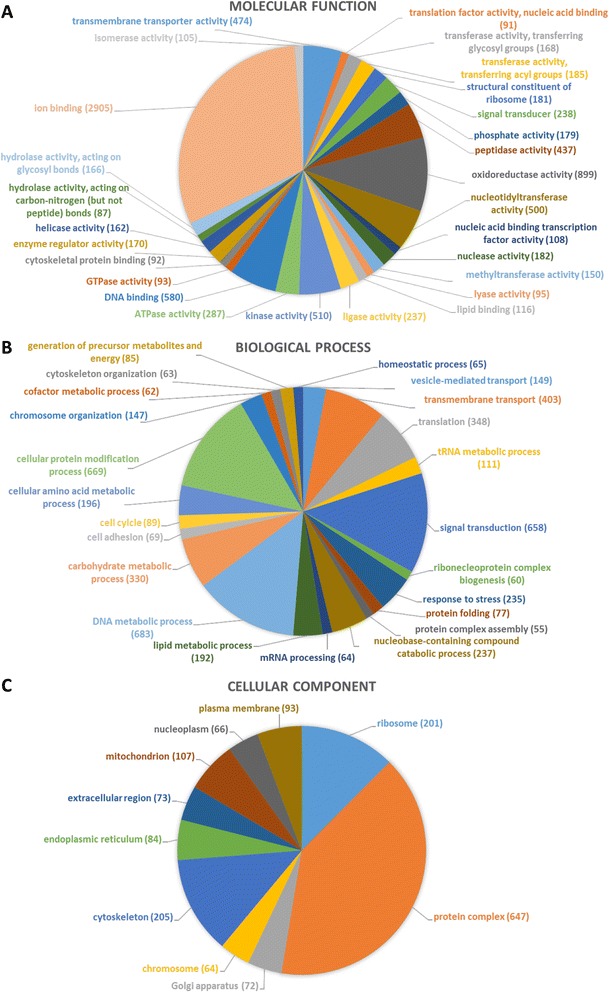
The de novo assembly of 1 billion DNA sequences to a reference genome of 393 Mb length provides an unprecedented insight into the I. ricinus genome. A homology search revealed sequences in the assembled genome contigs homologous to 89% of the I. scapularis genome scaffolds indicating coverage of most genome regions. We identified moreover 6,415 putative genes. More than 10,000 transcripts from naïve midgut were annotated with respect of predicted function and/or cellular localization. By combining an alignment-based with a motif-search-based annotation approach, we doubled the number of annotations throughout all functional categories. In addition, 574 gel spots were significantly identified by mass spectrometry (p<0.05) and 285 distinct proteins expressed in the naïve midgut were annotated functionally and/or for cellular localization. Our systems approach reveals a midgut metabolism of the unfed tick that is prepared to sense and process an anticipated blood meal.
This multiple-omics study vastly extends the publicly available DNA and RNA databases for I. ricinus, paving the way for further in-depth analysis of the most important European disease vector and its interactions with pathogens and hosts.
在欧洲,蓖麻硬蜱是威胁人类、家畜、野生动物和伴侣动物的疾病的最重要传播媒介。然而,其基因组序列信息缺失,转录本和蛋白质的功能注释有限。这种信息缺失限制了对该传播媒介及其与病原体和宿主相互作用的研究。在此,我们展示并整合了对蓖麻硬蜱基因组与未进食的蓖麻硬蜱中肠转录组和蛋白质组的首次分析。
对蓖麻硬蜱进行全基因组测序,并对序列进行从头组装。同时,解剖蓖麻硬蜱并对中肠转录组进行测序。通过转录本发现分析整合这两个数据集,以鉴定推定基因,并筛选基因组重叠群的同源性。基于比对和基于基序搜索的方法相结合用于中肠转录组的注释。此外,以公共数据库和内部构建的转录组数据库为参考,通过质谱鉴定和注释中肠蛋白质,并对结果进行交叉验证。
将10亿条DNA序列从头组装成一个长度为393 Mb的参考基因组,为深入了解蓖麻硬蜱基因组提供了前所未有的视角。同源性搜索显示,组装后的基因组重叠群中的序列与肩突硬蜱基因组支架的89%同源,表明覆盖了大多数基因组区域。我们还鉴定出6415个推定基因。根据预测功能和/或细胞定位,对来自未进食中肠的10000多个转录本进行了注释。通过将基于比对的注释方法与基于基序搜索的注释方法相结合,我们在所有功能类别中使注释数量增加了一倍。此外,通过质谱显著鉴定出574个凝胶点(p<0.05),并对未进食中肠中表达的285种不同蛋白质进行了功能和/或细胞定位注释。我们的系统方法揭示了未进食蜱的中肠代谢,该代谢准备感知和处理预期的血餐。
这项多组学研究极大地扩展了蓖麻硬蜱的公开DNA和RNA数据库,为进一步深入分析欧洲最重要的疾病传播媒介及其与病原体和宿主的相互作用铺平了道路。