Shi Shuhua, Zhang Shaolong, Zhang Qinggang
School of Science, Shandong Jianzhu University, Jinan, China.
College of Physics and Electronics, Shandong Normal University, Jinan, China.
PLoS One. 2015 Oct 29;10(10):e0141409. doi: 10.1371/journal.pone.0141409. eCollection 2015.
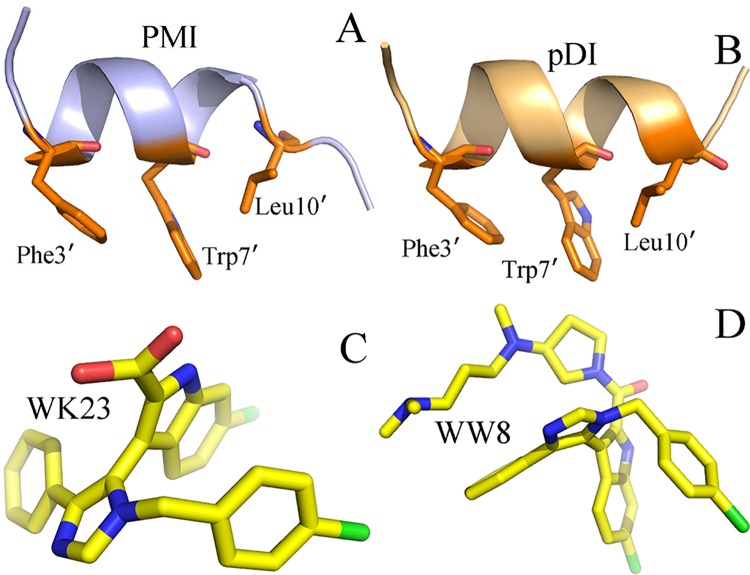
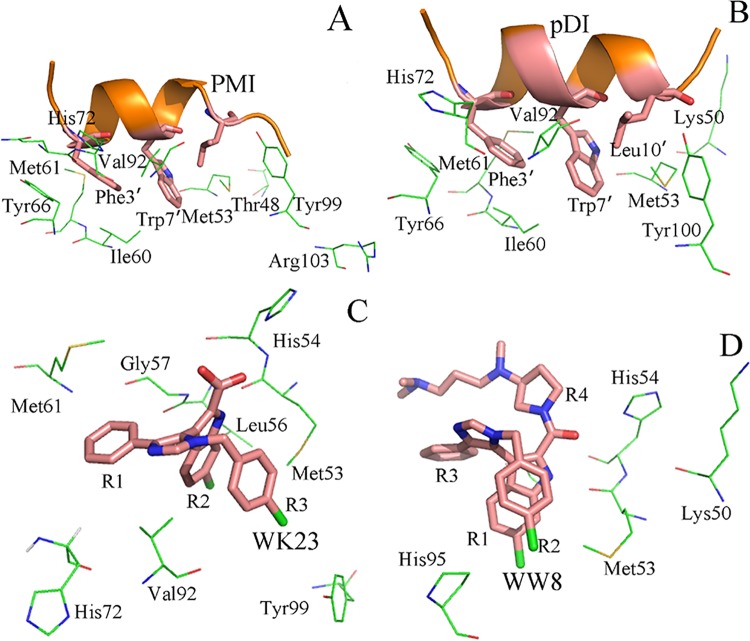
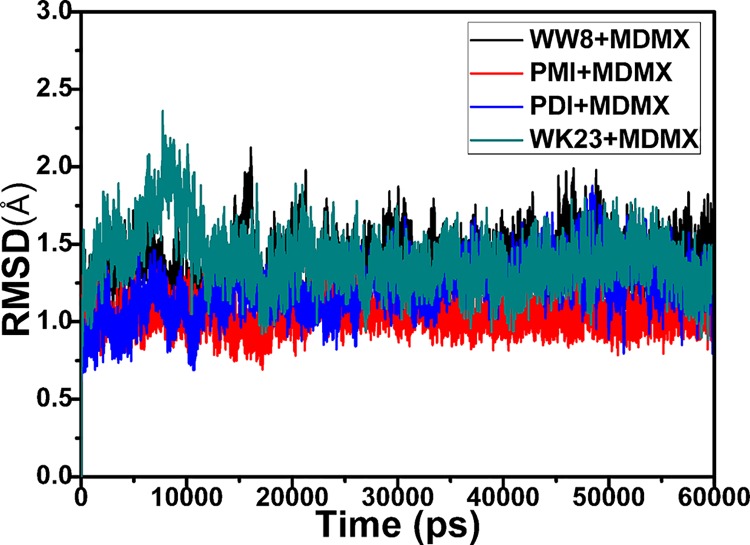
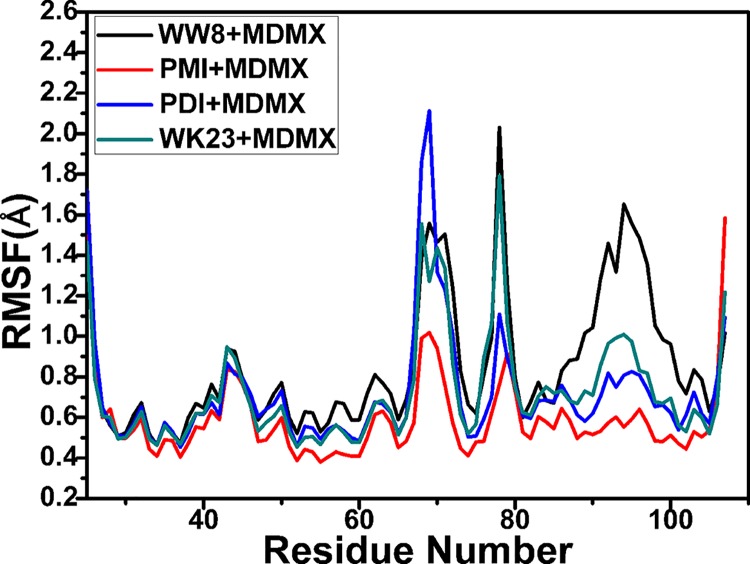
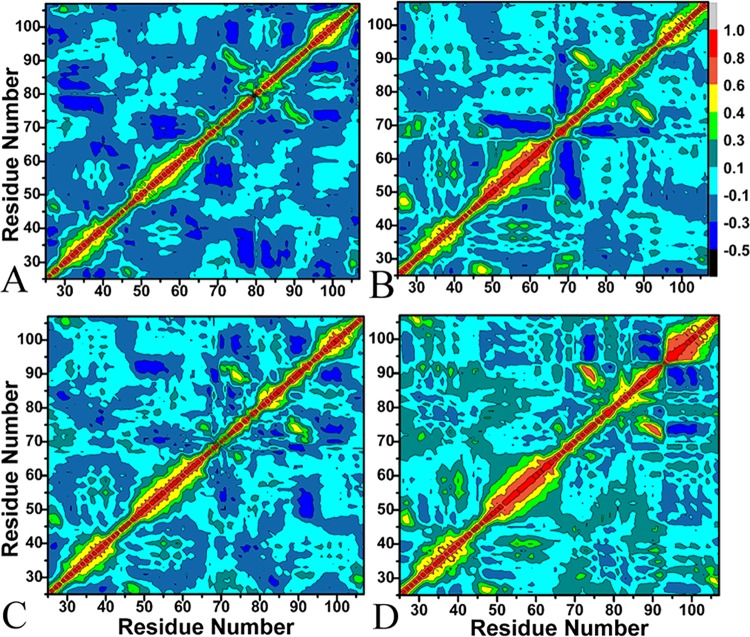
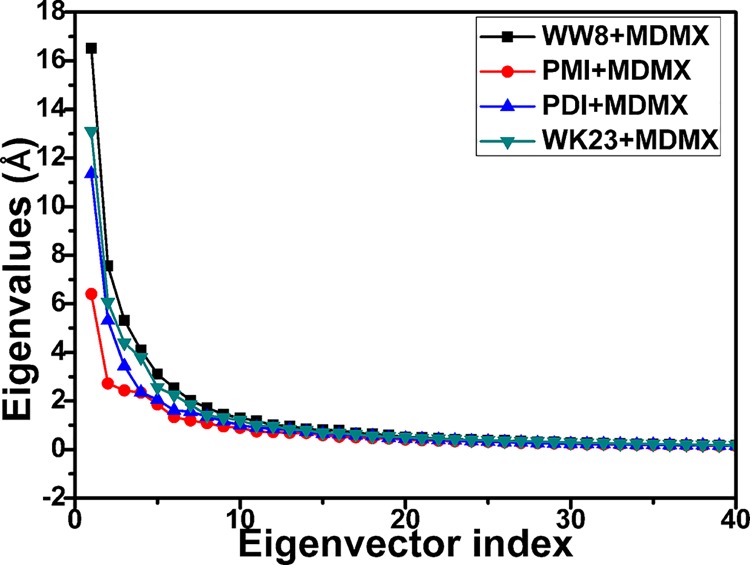
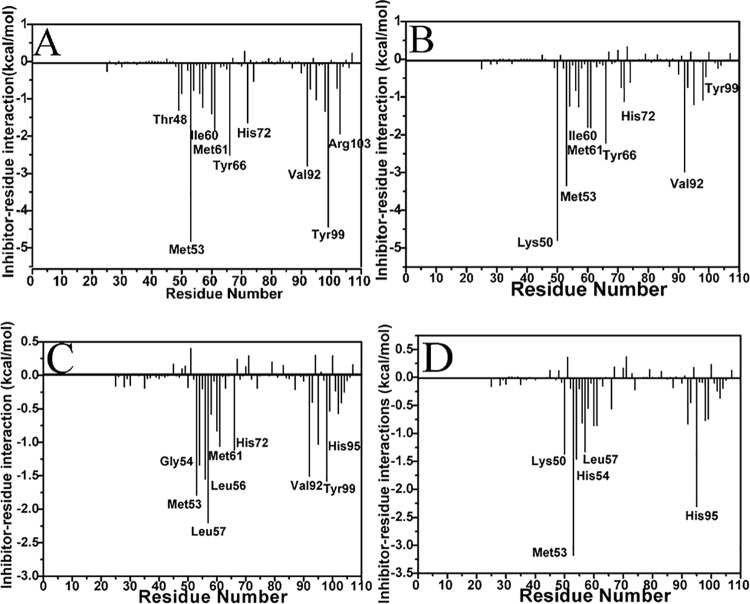
The p53-MDMX interaction has attracted extensive attention of anti-cancer drug development in recent years. This current work adopted molecular dynamics (MD) simulations and cross-correlation analysis to investigate conformation changes of MDMX caused by inhibitor bindings. The obtained information indicates that the binding cleft of MDMX undergoes a large conformational change and the dynamic behavior of residues obviously change by the presence of different structural inhibitors. Two different methods of binding free energy predictions were employed to carry out a comparable insight into binding mechanisms of four inhibitors PMI, pDI, WK23 and WW8 to MDMX. The data show that the main factor controlling the inhibitor bindings to MDMX arises from van der Waals interactions. The binding free energies were further divided into contribution of each residue and the derived information gives a conclusion that the hydrophobic interactions, such as CH-CH, CH-π and π-π interactions, are responsible for the inhibitor associations with MDMX.
近年来,p53-MDMX相互作用已引起抗癌药物研发的广泛关注。当前这项工作采用分子动力学(MD)模拟和交叉相关分析来研究抑制剂结合引起的MDMX构象变化。所获得的信息表明,MDMX的结合裂隙经历了较大的构象变化,并且不同结构抑制剂的存在明显改变了残基的动态行为。采用两种不同的结合自由能预测方法,对四种抑制剂PMI、pDI、WK23和WW8与MDMX的结合机制进行了可比的深入研究。数据表明,控制抑制剂与MDMX结合的主要因素来自范德华相互作用。结合自由能进一步分为每个残基的贡献,所得信息得出结论,即诸如CH-CH、CH-π和π-π相互作用等疏水相互作用是抑制剂与MDMX结合的原因。