Valdivia Hugo O, Scholte Larissa L S, Oliveira Guilherme, Gabaldón Toni, Bartholomeu Daniella C
Laboratório de Imunologia e Genômica de Parasitos, Instituto de Ciências Biológicas, Universidade Federal de Minas Gerais, Av. Presidente Antonio Carlos, 6627 - Pampulha, Belo Horizonte, MG, 31270-901, Brazil.
Department of Parasitology, U.S. Naval Medical Research Unit No. 6, Lima, Peru.
BMC Genomics. 2015 Oct 30;16:887. doi: 10.1186/s12864-015-2091-2.
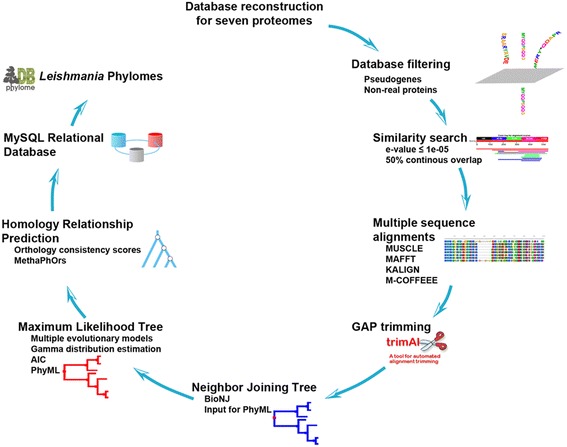
Leishmaniasis is a neglected parasitic disease with diverse clinical manifestations and a complex epidemiology. It has been shown that its parasite-related traits vary between species and that they modulate infectivity, pathogenicity, and virulence. However, understanding of the species-specific adaptations responsible for these features and their evolutionary background is limited. To improve our knowledge regarding the parasite biology and adaptation mechanisms of different Leishmania species, we conducted a proteome-wide phylogenomic analysis to gain insights into Leishmania evolution.
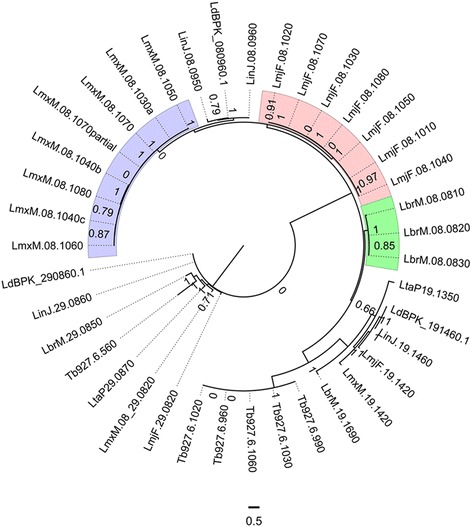
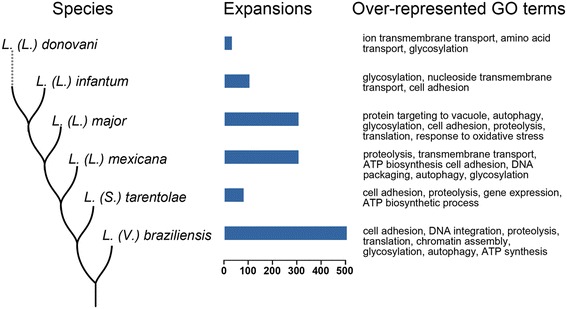
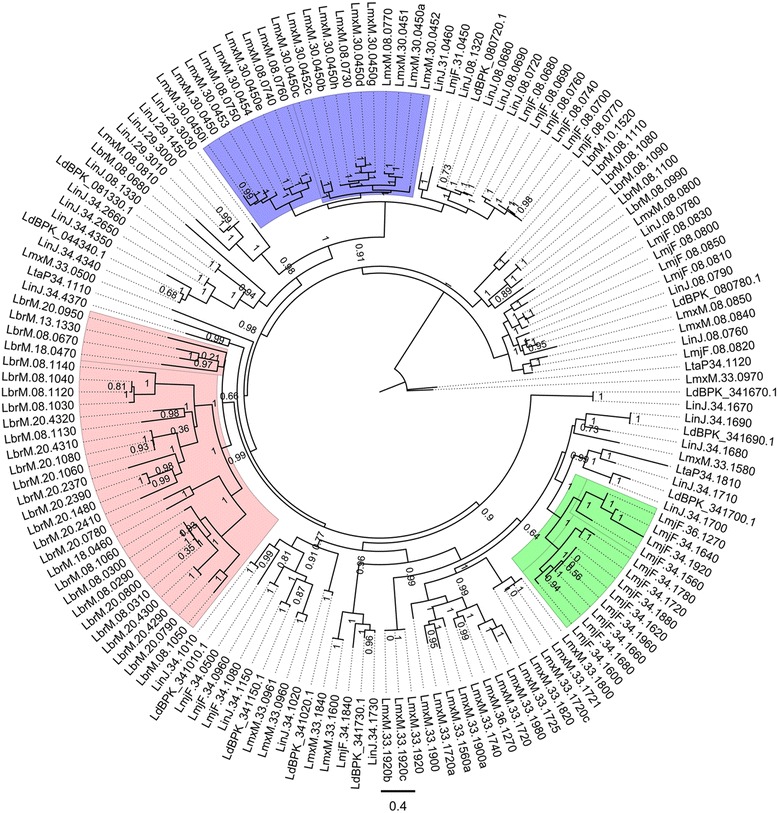
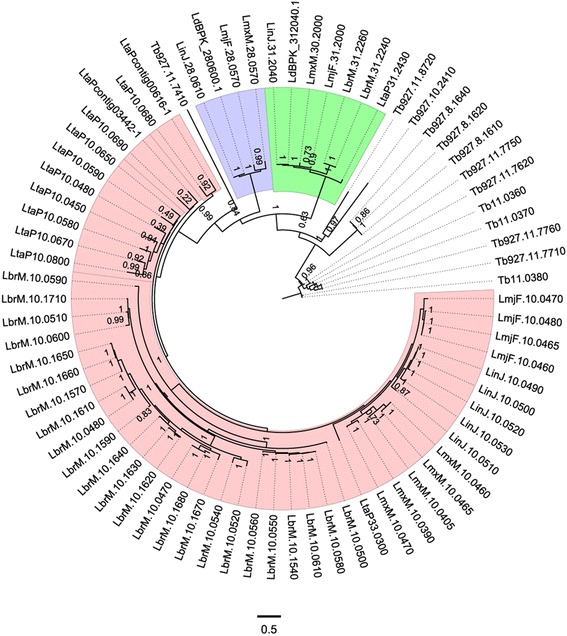
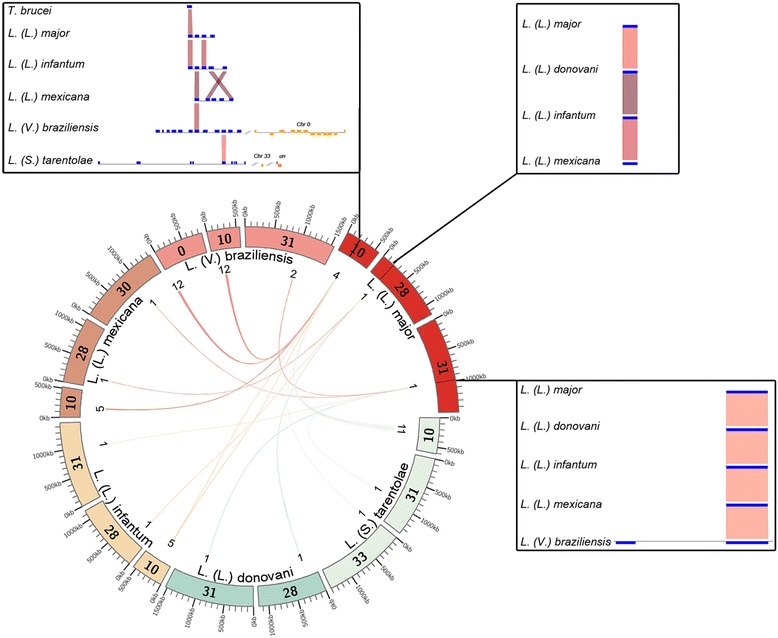
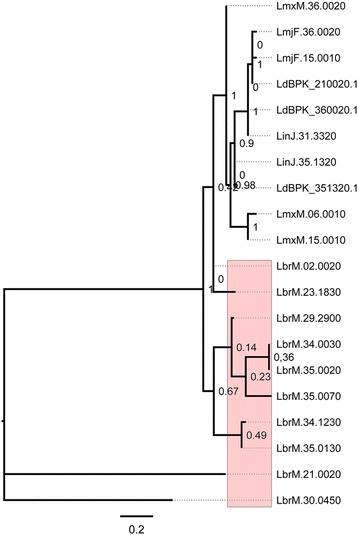
The analysis of the reconstructed phylomes (totaling 45,918 phylogenies) allowed us to detect genes that are shared in pathogenic Leishmania species, such as calpain-like cysteine peptidases and 3'a2rel-related proteins, or genes that could be associated with visceral or cutaneous development. This analysis also established the phylogenetic relationship of several hypothetical proteins whose roles remain to be characterized. Our findings demonstrated that gene duplication constitutes an important evolutionary force in Leishmania, acting on protein families that mediate host-parasite interactions, such as amastins, GP63 metallopeptidases, cathepsin L-like proteases, and our methods permitted a deeper analysis of their phylogenetic relationships.
Our results highlight the importance of proteome wide phylogenetic analyses to detect adaptation and evolutionary processes in different organisms and underscore the need to characterize the role of expanded and species-specific proteins in the context of Leishmania evolution by providing a framework for the phylogenetic relationships of Leishmania proteins. Phylogenomic data are publicly available for use through PhylomeDB (http://www.phylomedb.org).
利什曼病是一种被忽视的寄生虫病,临床表现多样,流行病学复杂。研究表明,其与寄生虫相关的特征在不同物种之间存在差异,并且这些特征会调节感染性、致病性和毒力。然而,对于导致这些特征的物种特异性适应性及其进化背景的了解仍然有限。为了增进我们对不同利什曼原虫物种的寄生虫生物学和适应机制的认识,我们进行了全蛋白质组系统发育基因组分析,以深入了解利什曼原虫的进化。
对重建的系统发育组(共45,918个系统发育树)的分析使我们能够检测到致病利什曼原虫物种中共享的基因,如钙蛋白酶样半胱氨酸肽酶和3'a2rel相关蛋白,或可能与内脏或皮肤发育相关的基因。该分析还确定了几种功能尚待确定的假设蛋白的系统发育关系。我们的研究结果表明,基因复制是利什曼原虫中的一种重要进化力量,作用于介导宿主 - 寄生虫相互作用的蛋白质家族,如无鞭毛体蛋白、GP63金属肽酶、组织蛋白酶L样蛋白酶,并且我们的方法允许对它们的系统发育关系进行更深入的分析。
我们的结果强调了全蛋白质组系统发育分析对于检测不同生物体中的适应性和进化过程的重要性,并强调需要通过提供利什曼原虫蛋白质系统发育关系的框架,在利什曼原虫进化的背景下确定扩展和物种特异性蛋白质的作用。系统发育基因组数据可通过PhylomeDB(http://www.phylomedb.org)公开获取以供使用。