Guo Jianjun, Fan Jue, Hauser Bernard A, Rhee Seung Y
Department of Plant Biology, Carnegie Institution for Science, Stanford, California 94305.
Department of Biology, Plant Molecular and Cellular Biology Program, University of Florida, Gainesville, Florida 32611.
G3 (Bethesda). 2015 Nov 3;6(1):67-77. doi: 10.1534/g3.115.023671.
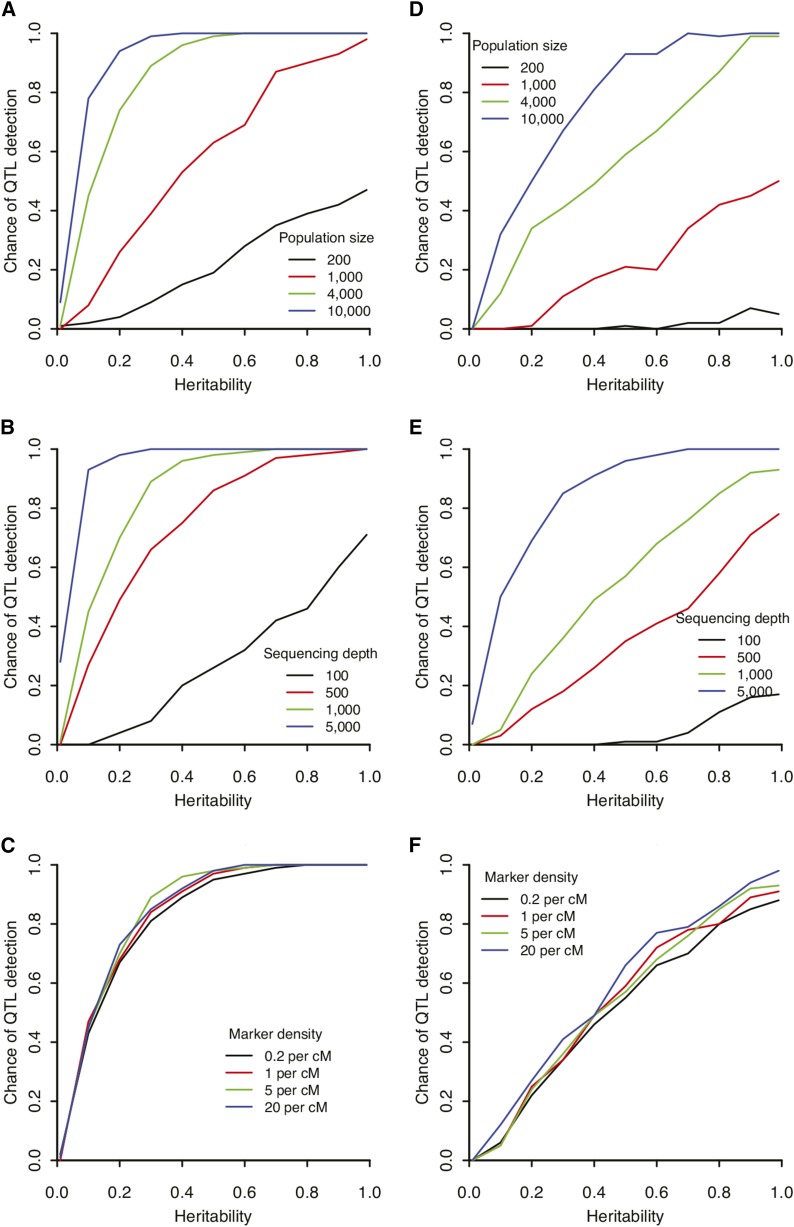
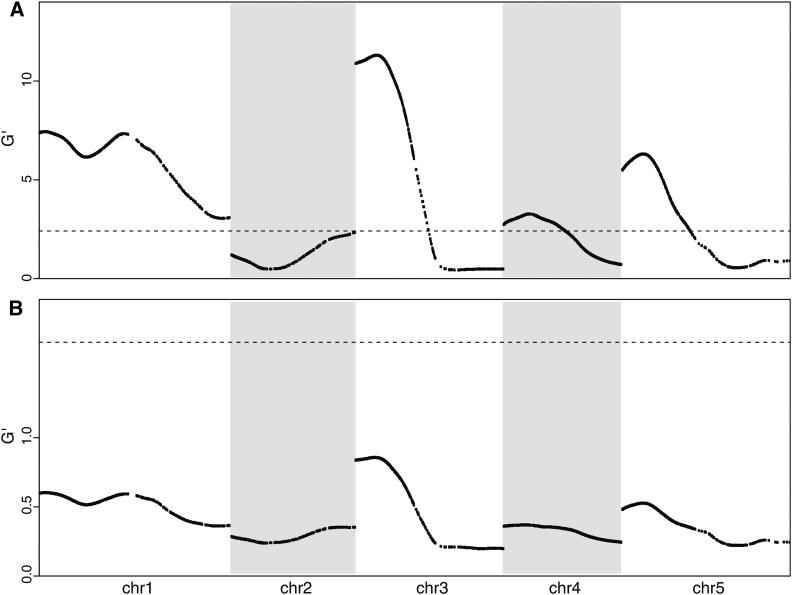
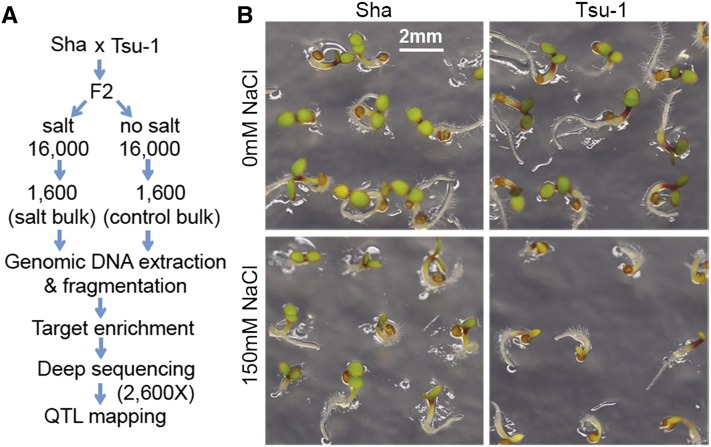
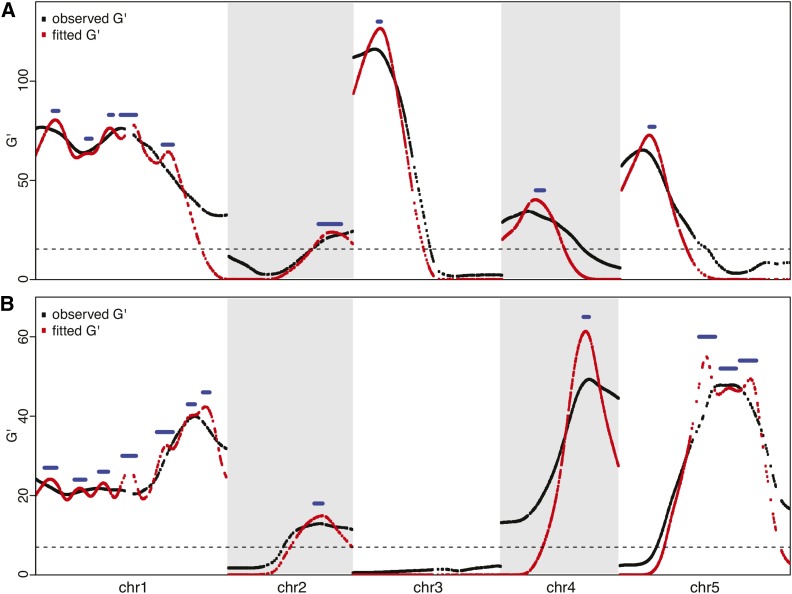
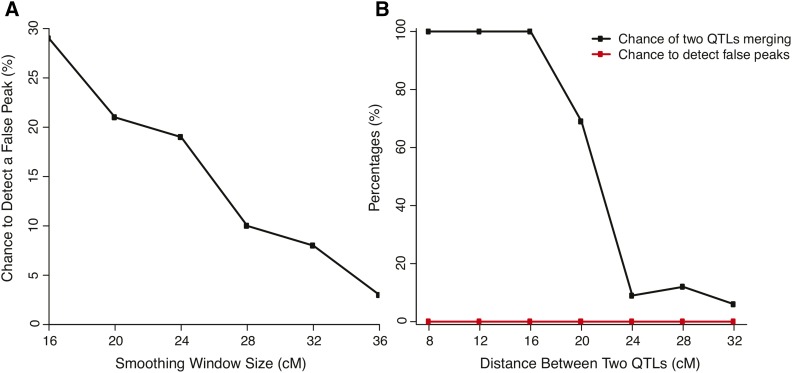
Complex traits such as crop performance and human diseases are controlled by multiple genetic loci, many of which have small effects and often go undetected by traditional quantitative trait locus (QTL) mapping. Recently, bulked segregant analysis with large F2 pools and genome-level markers (named extreme-QTL or X-QTL mapping) has been used to identify many QTL. To estimate parameters impacting QTL detection for X-QTL mapping, we simulated the effects of population size, marker density, and sequencing depth of markers on QTL detectability for traits with differing heritabilities. These simulations indicate that a high (>90%) chance of detecting QTL with at least 5% effect requires 5000× sequencing depth for a trait with heritability of 0.4-0.7. For most eukaryotic organisms, whole-genome sequencing at this depth is not economically feasible. Therefore, we tested and confirmed the feasibility of applying deep sequencing of target-enriched markers for X-QTL mapping. We used two traits in Arabidopsis thaliana with different heritabilities: seed size (H(2) = 0.61) and seedling greening in response to salt (H(2) = 0.94). We used a modified G test to identify QTL regions and developed a model-based statistical framework to resolve individual peaks by incorporating recombination rates. Multiple QTL were identified for both traits, including previously undiscovered QTL. We call our method target-enriched X-QTL (TEX-QTL) mapping; this mapping approach is not limited by the genome size or the availability of recombinant inbred populations and should be applicable to many organisms and traits.
诸如作物表现和人类疾病等复杂性状是由多个基因位点控制的,其中许多位点的效应较小,传统的数量性状位点(QTL)定位往往无法检测到。最近,利用大型F2群体和基因组水平标记的混合分离分析(称为极端QTL或X-QTL定位)已被用于鉴定许多QTL。为了估计影响X-QTL定位中QTL检测的参数,我们模拟了群体大小、标记密度和标记测序深度对不同遗传力性状的QTL可检测性的影响。这些模拟表明,对于遗传力为0.4-0.7的性状,检测效应至少为5%的QTL的概率要达到90%以上,需要5000倍的测序深度。对于大多数真核生物来说,在这个深度进行全基因组测序在经济上是不可行的。因此,我们测试并证实了将目标富集标记的深度测序应用于X-QTL定位的可行性。我们在拟南芥中使用了两个具有不同遗传力的性状:种子大小(H(2)=0.61)和对盐胁迫的幼苗绿化(H(2)=0.94)。我们使用改良的G检验来鉴定QTL区域,并开发了一个基于模型的统计框架,通过纳入重组率来解析单个峰值。两个性状都鉴定出了多个QTL,包括以前未发现的QTL。我们将我们的方法称为目标富集X-QTL(TEX-QTL)定位;这种定位方法不受基因组大小或重组自交群体可用性的限制,应该适用于许多生物和性状。