Röst Hannes L, Malmström Lars, Aebersold Ruedi
Department of Biology, Institute of Molecular Systems Biology, ETH Zurich, CH-8093 Zurich, Switzerland Department of Genetics, Stanford University, Stanford, CA 94305.
Department of Biology, Institute of Molecular Systems Biology, ETH Zurich, CH-8093 Zurich, Switzerland S3IT, University of Zurich, CH-8057 Zurich, Switzerland.
Mol Biol Cell. 2015 Nov 5;26(22):3926-31. doi: 10.1091/mbc.E15-07-0507.
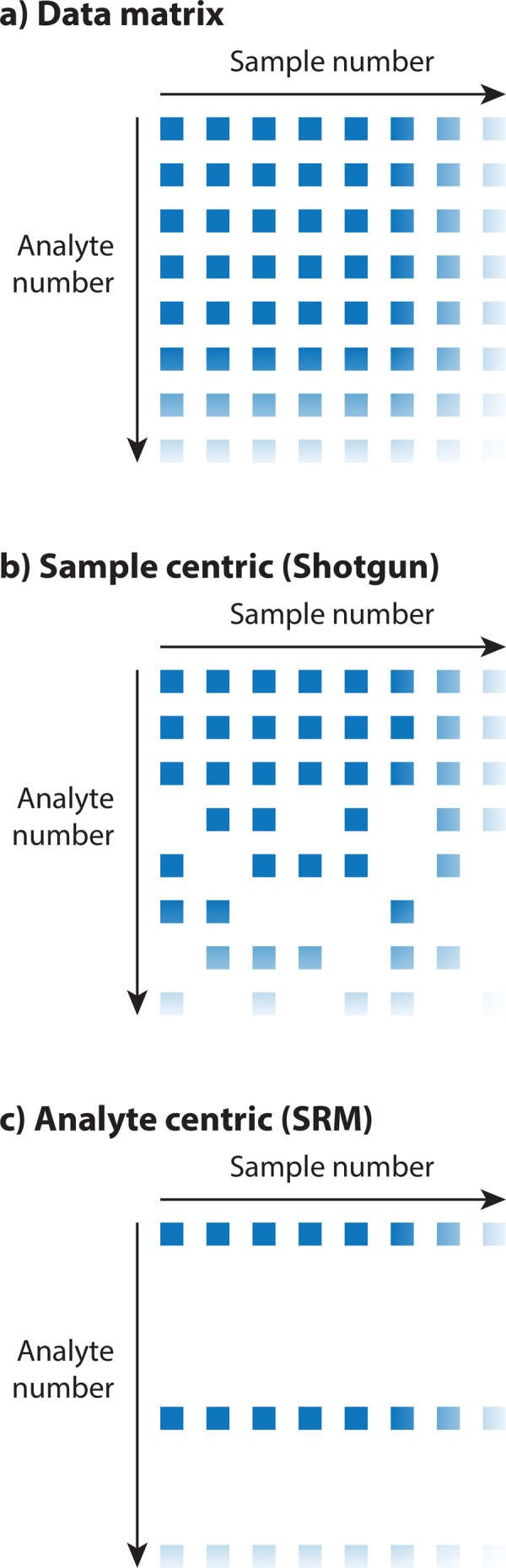
Historically, many mass spectrometry-based proteomic studies have aimed at compiling an inventory of protein compounds present in a biological sample, with the long-term objective of creating a proteome map of a species. However, to answer fundamental questions about the behavior of biological systems at the protein level, accurate and unbiased quantitative data are required in addition to a list of all protein components. Fueled by advances in mass spectrometry, the proteomics field has thus recently shifted focus toward the reproducible quantification of proteins across a large number of biological samples. This provides the foundation to move away from pure enumeration of identified proteins toward quantitative matrices of many proteins measured across multiple samples. It is argued here that data matrices consisting of highly reproducible, quantitative, and unbiased proteomic measurements across a high number of conditions, referred to here as quantitative proteotype maps, will become the fundamental currency in the field and provide the starting point for downstream biological analysis. Such proteotype data matrices, for example, are generated by the measurement of large patient cohorts, time series, or multiple experimental perturbations. They are expected to have a large effect on systems biology and personalized medicine approaches that investigate the dynamic behavior of biological systems across multiple perturbations, time points, and individuals.
从历史上看,许多基于质谱的蛋白质组学研究旨在编制生物样品中存在的蛋白质化合物清单,其长期目标是创建一个物种的蛋白质组图谱。然而,要回答关于生物系统在蛋白质水平上行为的基本问题,除了所有蛋白质成分的列表外,还需要准确且无偏差的定量数据。受质谱技术进步的推动,蛋白质组学领域最近因此将重点转向了对大量生物样品中蛋白质的可重复定量。这为从单纯列举已鉴定蛋白质转向跨多个样品测量的多种蛋白质的定量矩阵奠定了基础。本文认为,由在大量条件下高度可重复、定量且无偏差的蛋白质组学测量组成的数据矩阵,在此称为定量蛋白质型图谱,将成为该领域的基本货币,并为下游生物学分析提供起点。例如,此类蛋白质型数据矩阵是通过对大量患者队列、时间序列或多次实验扰动的测量生成的。它们预计会对研究生物系统在多种扰动、时间点和个体间动态行为的系统生物学和个性化医学方法产生重大影响。