Salovska Barbora, Zhu Hongwen, Gandhi Tejas, Frank Max, Li Wenxue, Rosenberger George, Wu Chongde, Germain Pierre-Luc, Zhou Hu, Hodny Zdenek, Reiter Lukas, Liu Yansheng
Yale Cancer Biology Institute, Yale University, West Haven, CT, USA.
Department of Genome Integrity, Institute of Molecular Genetics of the Czech Academy of Sciences, Prague, Czech Republic.
Mol Syst Biol. 2020 Mar;16(3):e9170. doi: 10.15252/msb.20199170.
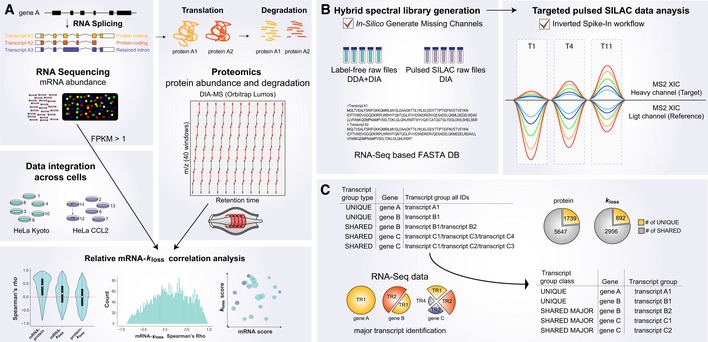
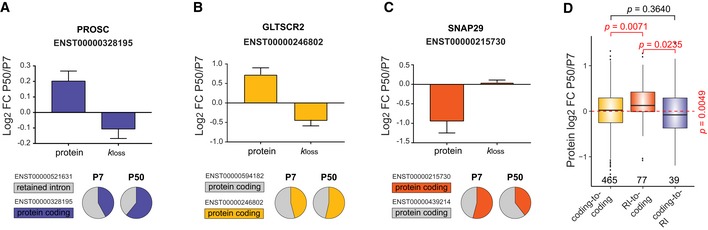
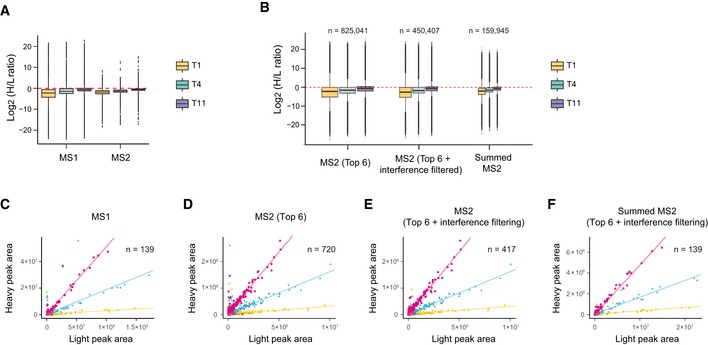
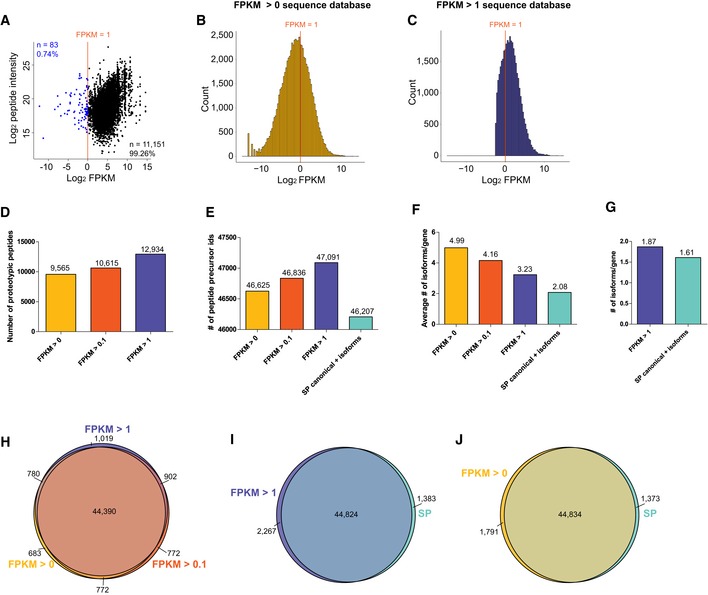
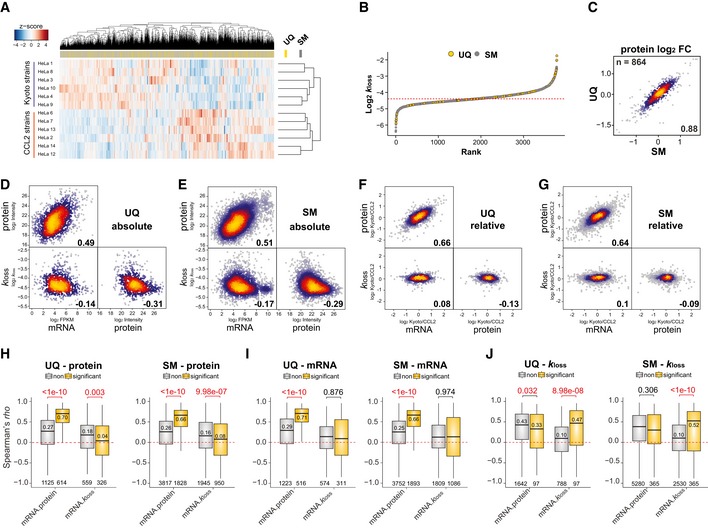
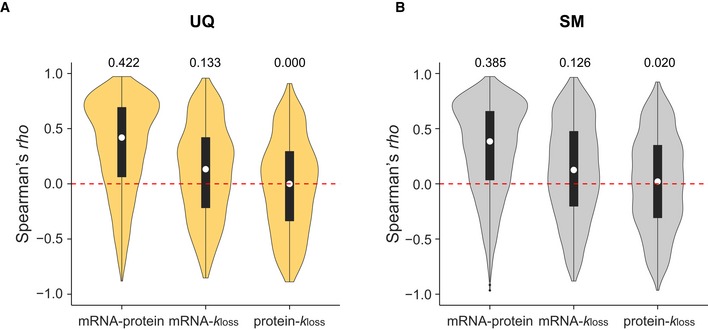
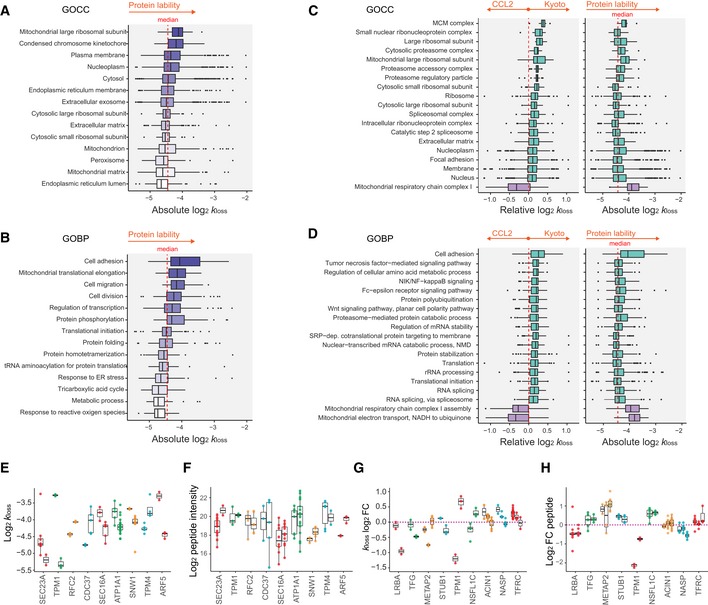
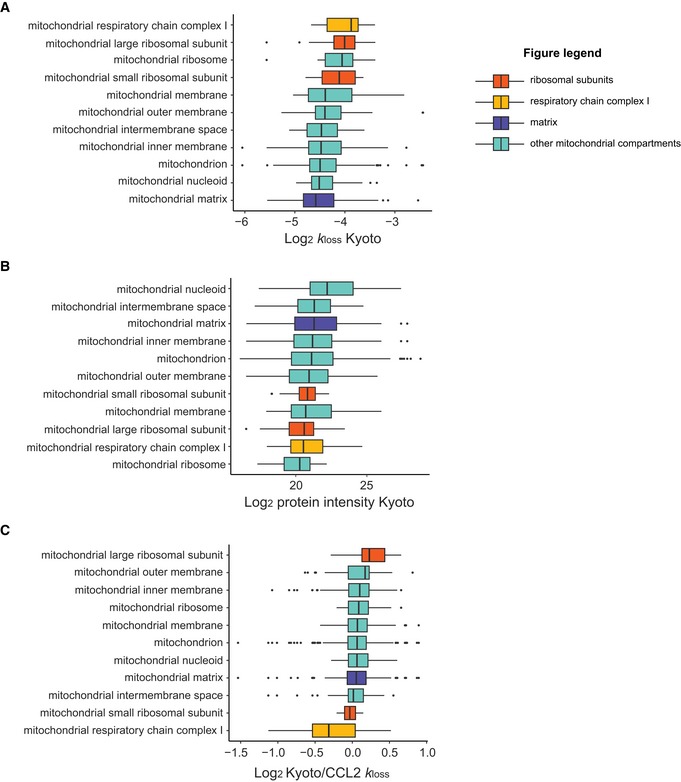
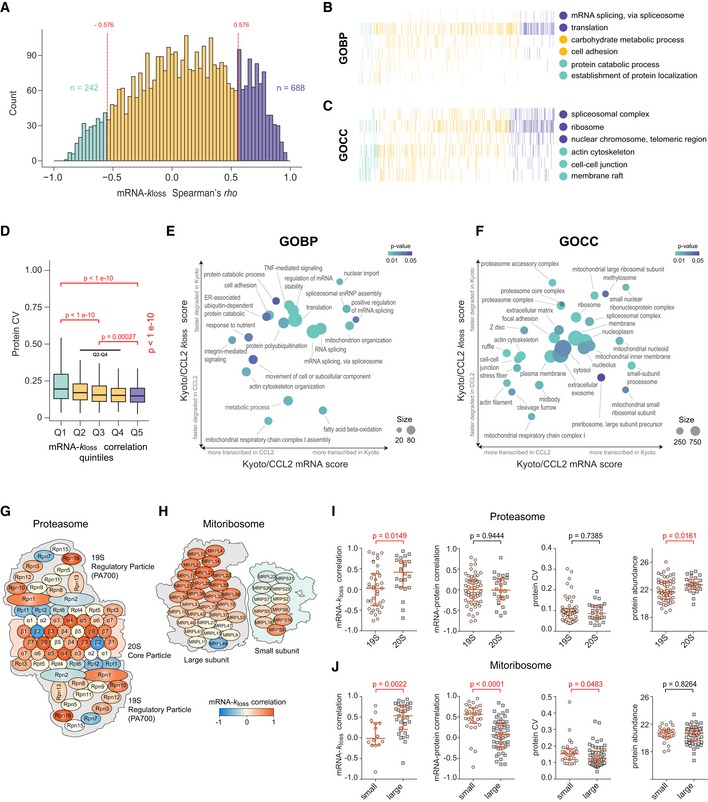
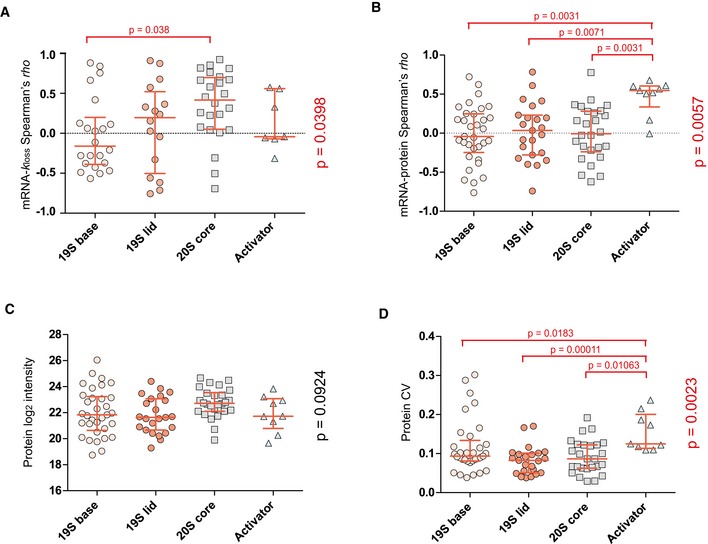
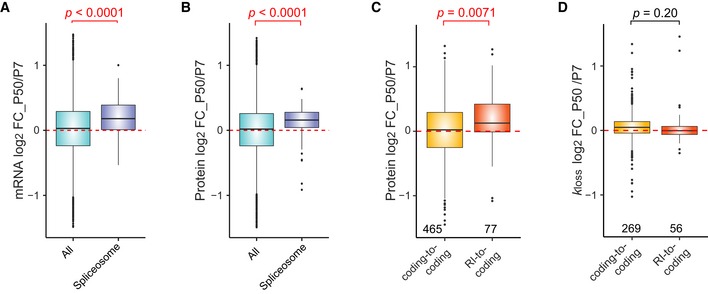
Profiling of biological relationships between different molecular layers dissects regulatory mechanisms that ultimately determine cellular function. To thoroughly assess the role of protein post-translational turnover, we devised a strategy combining pulse stable isotope-labeled amino acids in cells (pSILAC), data-independent acquisition mass spectrometry (DIA-MS), and a novel data analysis framework that resolves protein degradation rate on the level of mRNA alternative splicing isoforms and isoform groups. We demonstrated our approach by the genome-wide correlation analysis between mRNA amounts and protein degradation across different strains of HeLa cells that harbor a high grade of gene dosage variation. The dataset revealed that specific biological processes, cellular organelles, spatial compartments of organelles, and individual protein isoforms of the same genes could have distinctive degradation rate. The protein degradation diversity thus dissects the corresponding buffering or concerting protein turnover control across cancer cell lines. The data further indicate that specific mRNA splicing events such as intron retention significantly impact the protein abundance levels. Our findings support the tight association between transcriptome variability and proteostasis and provide a methodological foundation for studying functional protein degradation.
剖析不同分子层之间的生物学关系,有助于揭示最终决定细胞功能的调控机制。为了全面评估蛋白质翻译后周转的作用,我们设计了一种策略,将细胞脉冲稳定同位素标记氨基酸(pSILAC)、数据非依赖采集质谱(DIA-MS)以及一种新型数据分析框架相结合,该框架可在mRNA可变剪接异构体和异构体组水平上解析蛋白质降解速率。我们通过对不同HeLa细胞株进行全基因组相关性分析来验证我们的方法,这些细胞株具有高度的基因剂量变异。数据集显示,特定的生物学过程、细胞器、细胞器的空间区室以及同一基因的单个蛋白质异构体可能具有不同的降解速率。因此,蛋白质降解的多样性揭示了癌细胞系中相应的缓冲或协同蛋白质周转控制。数据还进一步表明,特定的mRNA剪接事件,如内含子保留,会显著影响蛋白质丰度水平。我们的研究结果支持转录组变异性与蛋白质稳态之间的紧密关联,并为研究功能性蛋白质降解提供了方法学基础。