Hoeper Marius M, Behr Juergen, Held Matthias, Grunig Ekkehard, Vizza C Dario, Vonk-Noordegraaf Anton, Lange Tobias J, Claussen Martin, Grohé Christian, Klose Hans, Olsson Karen M, Zelniker Thomas, Neurohr Claus, Distler Oliver, Wirtz Hubert, Opitz Christian, Huscher Doerte, Pittrow David, Gibbs J Simon R
Department of Respiratory Medicine and German Center of Lung Research (DZL), Hannover Medical School, Hannover, Germany.
Department of Internal Medicine V, University of Munich, Munich, Germany.
PLoS One. 2015 Dec 2;10(12):e0141911. doi: 10.1371/journal.pone.0141911. eCollection 2015.
Pulmonary hypertension (PH) is a common finding in patients with chronic fibrosing idiopathic interstitial pneumonias (IIP). Little is known about the response to pulmonary vasodilator therapy in this patient population. COMPERA is an international registry that prospectively captures data from patients with various forms of PH receiving pulmonary vasodilator therapies.
We retrieved data from COMPERA to compare patient characteristics, treatment patterns, response to therapy and survival in newly diagnosed patients with idiopathic pulmonary arterial hypertension (IPAH) and PH associated with IIP (PH-IIP).
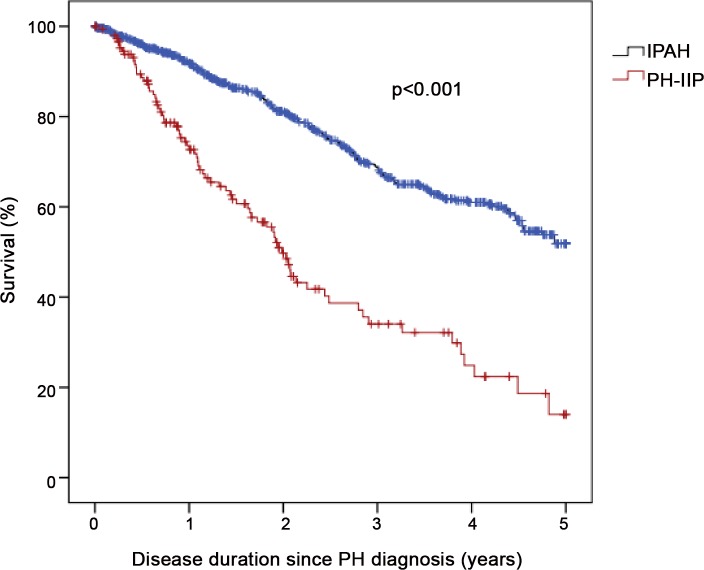
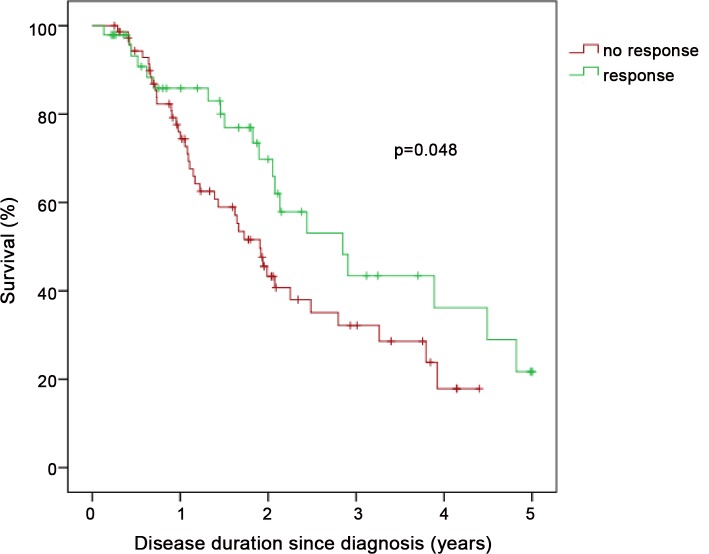
Compared to patients with IPAH (n = 798), patients with PH-IIP (n = 151) were older and predominantly males. Patients with PH-IIP were treated predominantly with phosphodiesterase-5 inhibitors (88% at entry, 87% after 1 year). From baseline to the first follow-up visit, the median improvement in 6MWD was 30 m in patients with IPAH and 24.5 m in patients with PH-IIP (p = 0.457 for the difference between both groups). Improvements in NYHA functional class were observed in 22.4% and 29.5% of these patients, respectively (p = 0.179 for the difference between both groups). Survival rates were significantly worse in PH-IIP than in IPAH (3-year survival 34.0 versus 68.6%; p<0.001). Total lung capacity, NYHA class IV, and mixed-venous oxygen saturation were independent predictors of survival in patients with PH-IIP.
Patients with PH-IIP have a dismal prognosis. Our results suggest that pulmonary vasodilator therapy may be associated with short-term functional improvement in some of these patients but it is unclear whether this treatment affects survival.
clinicaltrials.gov NCT01347216.
肺动脉高压(PH)在慢性纤维化性特发性间质性肺炎(IIP)患者中很常见。对于该患者群体对肺血管扩张剂治疗的反应了解甚少。COMPERA是一个国际注册机构,前瞻性地收集接受肺血管扩张剂治疗的各种形式PH患者的数据。
我们从COMPERA检索数据,以比较新诊断的特发性肺动脉高压(IPAH)和与IIP相关的PH(PH-IIP)患者的特征、治疗模式、治疗反应和生存率。
与IPAH患者(n = 798)相比,PH-IIP患者(n = 151)年龄更大,且男性居多。PH-IIP患者主要接受磷酸二酯酶-5抑制剂治疗(入组时88%,1年后87%)。从基线到首次随访,IPAH患者6分钟步行距离(6MWD)的中位数改善为30米,PH-IIP患者为24.5米(两组间差异p = 0.457)。这些患者中分别有22.4%和29.5%的纽约心脏协会(NYHA)功能分级得到改善(两组间差异p = 0.179)。PH-IIP患者的生存率明显低于IPAH患者(3年生存率分别为34.0%和68.6%;p<0.001)。肺总量、NYHA IV级和混合静脉血氧饱和度是PH-IIP患者生存的独立预测因素。
PH-IIP患者预后不佳。我们的结果表明,肺血管扩张剂治疗可能与部分此类患者的短期功能改善有关,但尚不清楚这种治疗是否影响生存。
clinicaltrials.gov NCT01347216