Ramazzotti Matteo, Berná Luisa, Donati Claudio, Cavalieri Duccio
Dipartimento di Scienze Biomediche Sperimentali e Cliniche, Università degli Studi di Firenze Firenze, Italy.
Unidad de Biología Molecular, Institut Pasteur de Montevideo Montevideo, Uruguay.
Front Genet. 2015 Nov 17;6:329. doi: 10.3389/fgene.2015.00329. eCollection 2015.
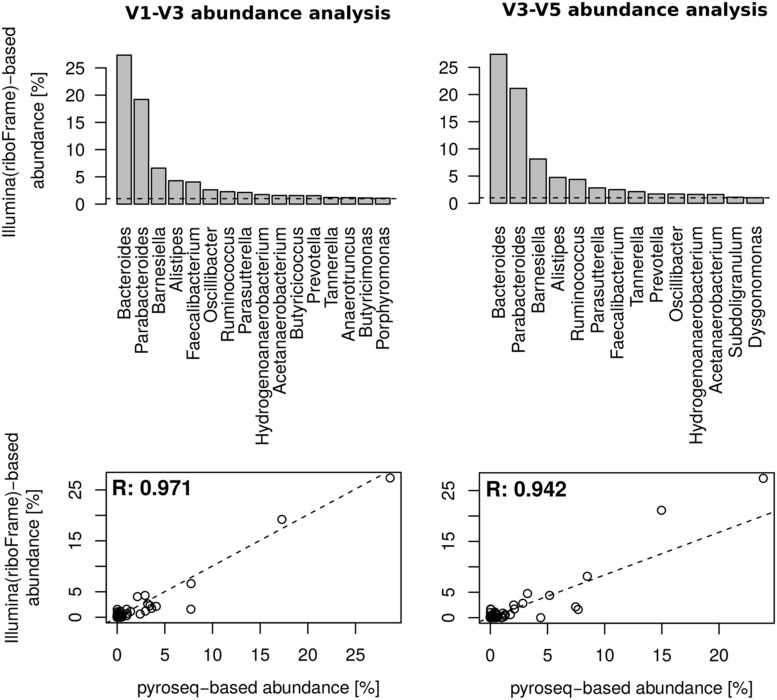
Non-targeted metagenomics offers the unprecedented possibility of simultaneously investigate the microbial profile and the genetic capabilities of a sample by a direct analysis of its entire DNA content. The assessment of the microbial taxonomic composition is frequently obtained by mapping reads to genomic databases that, although growing, are still limited and biased. Here we present riboFrame, a novel procedure for microbial profiling based on the identification and classification of 16S rDNA sequences in non-targeted metagenomics datasets. Reads overlapping the 16S rDNA genes are identified using Hidden Markov Models and a taxonomic assignment is obtained by naïve Bayesian classification. All reads identified as ribosomal are coherently positioned in the 16S rDNA gene, allowing the use of the topology of the gene (i.e., the secondary structure and the location of variable regions) to guide the abundance analysis. We tested and verified the effectiveness of our method on simulated ribosomal data, on simulated metagenomes and on a real dataset. riboFrame exploits the taxonomic potentialities of the 16S rDNA gene in the context of non-targeted metagenomics, giving an accurate perspective on the microbial profile in metagenomic samples.
非靶向宏基因组学提供了前所未有的可能性,即通过直接分析样本的全部DNA含量,同时研究其微生物概况和遗传能力。微生物分类组成的评估通常是通过将 reads 映射到基因组数据库来实现的,尽管这些数据库在不断增长,但仍然有限且存在偏差。在这里,我们提出了 riboFrame,这是一种基于非靶向宏基因组学数据集中16S rDNA序列的鉴定和分类的微生物分析新方法。使用隐马尔可夫模型识别与16S rDNA基因重叠的 reads,并通过朴素贝叶斯分类获得分类学分配。所有被鉴定为核糖体的 reads 都被连贯地定位在16S rDNA基因中,从而可以利用基因的拓扑结构(即二级结构和可变区的位置)来指导丰度分析。我们在模拟核糖体数据、模拟宏基因组和真实数据集上测试并验证了我们方法的有效性。riboFrame在非靶向宏基因组学背景下利用了(16S) rDNA基因的分类学潜力,为宏基因组样本中的微生物概况提供了准确的视角。