Reuter Sandra, Török M Estée, Holden Matthew T G, Reynolds Rosy, Raven Kathy E, Blane Beth, Donker Tjibbe, Bentley Stephen D, Aanensen David M, Grundmann Hajo, Feil Edward J, Spratt Brian G, Parkhill Julian, Peacock Sharon J
Department of Medicine, University of Cambridge, Cambridge CB2 0QQ, United Kingdom; Pathogen Genomics, Wellcome Trust Sanger Institute, Hinxton CB10 1SA, United Kingdom;
Department of Medicine, University of Cambridge, Cambridge CB2 0QQ, United Kingdom; Public Health England, Microbiology Services Division, Addenbrooke's Hospital, Cambridge CB2 0QW, United Kingdom; Cambridge University Hospitals NHS Foundation Trust, Cambridge CB2 0QQ, United Kingdom;
Genome Res. 2016 Feb;26(2):263-70. doi: 10.1101/gr.196709.115. Epub 2015 Dec 15.
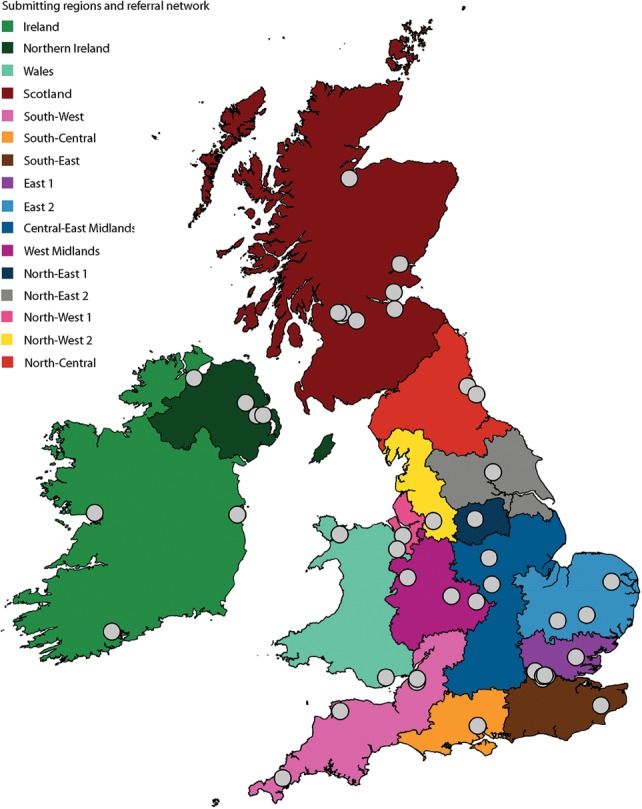
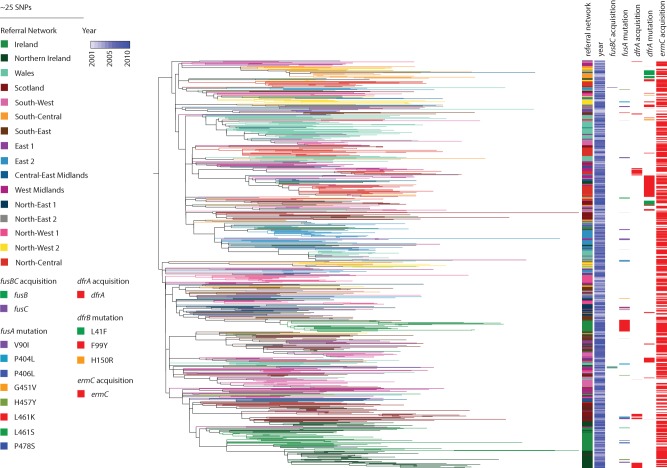
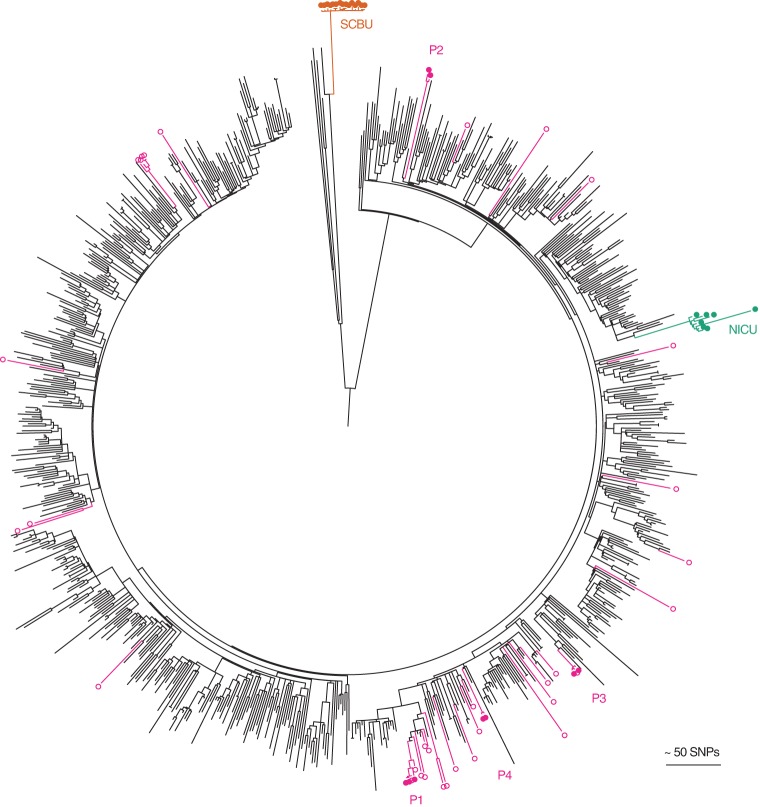
The correct interpretation of microbial sequencing data applied to surveillance and outbreak investigation depends on accessible genomic databases to provide vital genetic context. Our aim was to construct and describe a United Kingdom MRSA database containing over 1000 methicillin-resistant Staphylococcus aureus (MRSA) genomes drawn from England, Northern Ireland, Wales, Scotland, and the Republic of Ireland over a decade. We sequenced 1013 MRSA submitted to the British Society for Antimicrobial Chemotherapy by 46 laboratories between 2001 and 2010. Each isolate was assigned to a regional healthcare referral network in England and was otherwise grouped based on country of origin. Phylogenetic reconstructions were used to contextualize MRSA outbreak investigations and to detect the spread of resistance. The majority of isolates (n = 783, 77%) belonged to CC22, which contains the dominant United Kingdom epidemic clone (EMRSA-15). There was marked geographic structuring of EMRSA-15, consistent with widespread dissemination prior to the sampling decade followed by local diversification. The addition of MRSA genomes from two outbreaks and one pseudo-outbreak demonstrated the certainty with which outbreaks could be confirmed or refuted. We identified local and regional differences in antibiotic resistance profiles, with examples of local expansion, as well as widespread circulation of mobile genetic elements across the bacterial population. We have generated a resource for the future surveillance and outbreak investigation of MRSA in the United Kingdom and Ireland and have shown the value of this during outbreak investigation and tracking of antimicrobial resistance.
将微生物测序数据正确应用于监测和疫情调查,取决于是否有可获取的基因组数据库来提供重要的遗传背景信息。我们的目标是构建并描述一个英国耐甲氧西林金黄色葡萄球菌(MRSA)数据库,该数据库包含从英格兰、北爱尔兰、威尔士、苏格兰和爱尔兰共和国收集的1000多个MRSA基因组,时间跨度超过十年。我们对2001年至2010年间46个实验室提交给英国抗菌化疗学会的1013株MRSA进行了测序。每个分离株被分配到英格兰的一个区域医疗转诊网络,其他则根据原产国进行分组。系统发育重建用于将MRSA疫情调查置于背景中,并检测耐药性的传播。大多数分离株(n = 783,77%)属于CC22,其中包含英国主要的流行克隆(EMRSA - 15)。EMRSA - 15存在明显的地理结构,这与在采样十年之前广泛传播然后局部多样化一致。添加来自两次疫情和一次假疫情的MRSA基因组,证明了可以确定地证实或反驳疫情。我们确定了抗生素耐药谱的局部和区域差异,包括局部扩张的例子,以及移动遗传元件在细菌群体中的广泛传播。我们为英国和爱尔兰未来的MRSA监测和疫情调查生成了一个资源,并展示了其在疫情调查和抗菌药物耐药性追踪中的价值。