University of Cambridge, Department of Medicine, Cambridge, United Kingdom.
Wellcome Sanger Institute, Hinxton, United Kingdom.
Euro Surveill. 2019 Jan;24(4). doi: 10.2807/1560-7917.ES.2019.24.4.1800215.
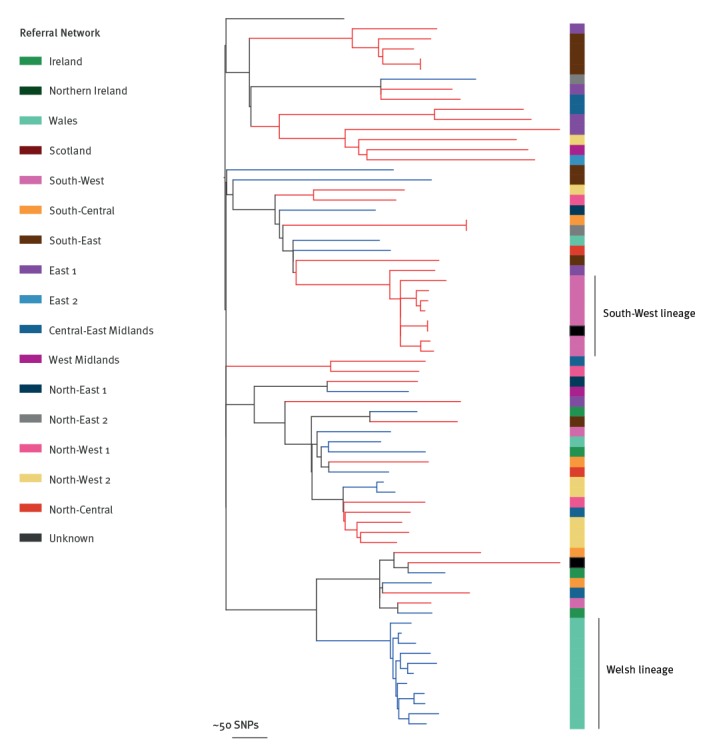
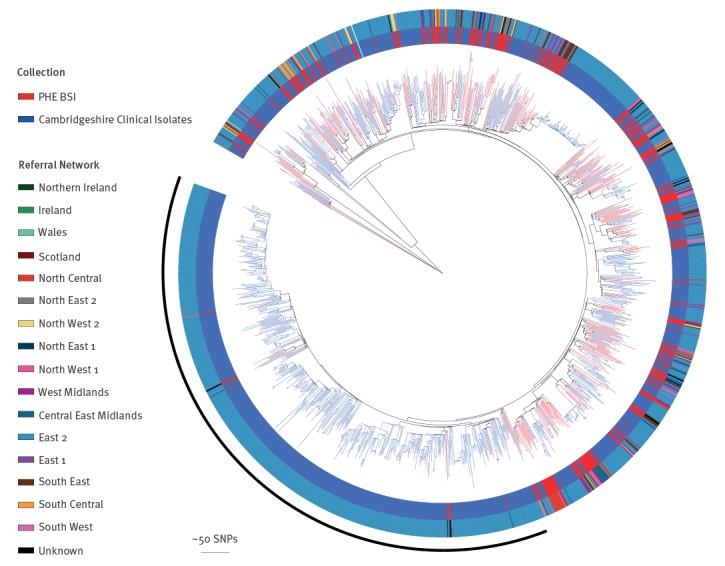
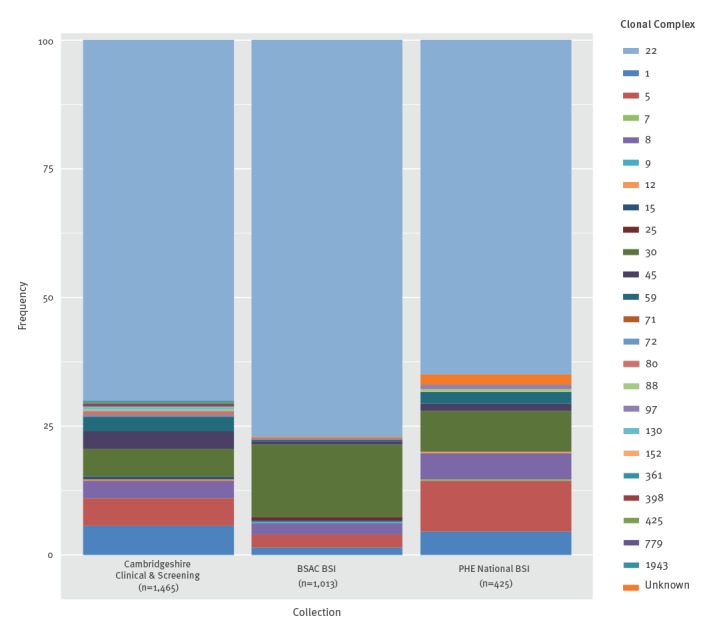
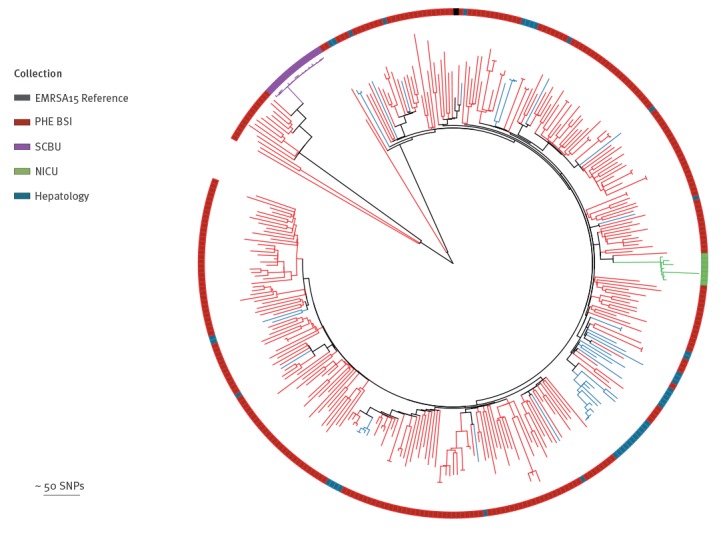
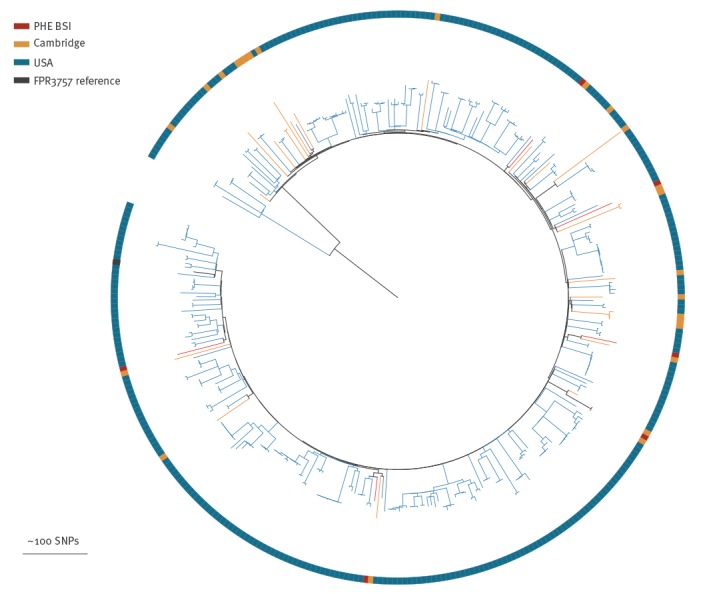
BackgroundMandatory reporting of methicillin-resistant (MRSA) bloodstream infections (BSI) has occurred in England for over 15years. Epidemiological information is recorded, but routine collection of isolates for characterisation has not been routinely undertaken. Ongoing developments in whole-genome sequencing (WGS) have demonstrated its value in outbreak investigations and for determining the spread of antimicrobial resistance and bacterial population structure. Benefits of adding genomics to routine epidemiological MRSA surveillance are unknown.AimTo determine feasibility and potential utility of adding genomics to epidemiological surveillance of MRSA.MethodsWe conducted an epidemiological and genomic survey of MRSA BSI in England over a 1-year period (1 October 2012--30 September 2013).ResultsDuring the study period, 903 cases of MRSA BSI were reported; 425 isolates were available for sequencing of which, 276 (65%) were clonal complex (CC) 22. Addition of 64 MRSA genomes from published outbreak investigations showed that the study genomes could provide context for outbreak isolates and supported cluster identification. Comparison to other MRSA genome collections demonstrated variation in clonal diversity achieved through different sampling strategies and identified potentially high-risk clones e.g. USA300 and local expansion of CC5 MRSA in South West England.ConclusionsWe demonstrate the potential utility of combined epidemiological and genomic MRSA BSI surveillance to determine the national population structure of MRSA, contextualise previous MRSA outbreaks, and detect potentially high-risk lineages. These findings support the integration of epidemiological and genomic surveillance for MRSA BSI as a step towards a comprehensive surveillance programme in England.
背景
在英格兰,耐甲氧西林金黄色葡萄球菌(MRSA)血流感染(BSI)的强制性报告已经实施了超过 15 年。已经记录了流行病学信息,但常规收集用于特征描述的分离株尚未常规进行。全基因组测序(WGS)的持续发展表明其在暴发调查以及确定抗生素耐药性和细菌种群结构传播方面具有价值。将基因组学添加到常规流行病学 MRSA 监测中的益处尚不清楚。
目的
确定将基因组学添加到 MRSA 流行病学监测中的可行性和潜在用途。
方法
我们在英格兰进行了为期一年(2012 年 10 月 1 日至 2013 年 9 月 30 日)的 MRSA BSI 流行病学和基因组调查。
结果
在研究期间,报告了 903 例 MRSA BSI 病例;有 425 株分离物可用于测序,其中 276 株(65%)为克隆复合体(CC)22。添加了来自已发表暴发调查的 64 株 MRSA 基因组,表明研究基因组可以为暴发分离物提供背景,并支持簇识别。与其他 MRSA 基因组集的比较表明,通过不同的采样策略实现了克隆多样性的变化,并确定了潜在的高风险克隆,例如 USA300 和英格兰西南部 CC5 MRSA 的局部扩张。
结论
我们证明了结合流行病学和基因组学的 MRSA BSI 监测在确定 MRSA 的全国种群结构、使先前的 MRSA 暴发具有背景以及检测潜在高风险谱系方面的潜在用途。这些发现支持将流行病学和基因组监测整合到 MRSA BSI 中,作为英格兰全面监测计划的一个步骤。