Ruffalo Matthew, Koyutürk Mehmet, Sharan Roded
Department of Electrical Engineering and Computer Science, Case Western Reserve University, Cleveland, Ohio, United States of America.
Center for Proteomics and Bioinformatics, Case Western Reserve University, Cleveland, Ohio, United States of America.
PLoS Comput Biol. 2015 Dec 18;11(12):e1004595. doi: 10.1371/journal.pcbi.1004595. eCollection 2015 Dec.
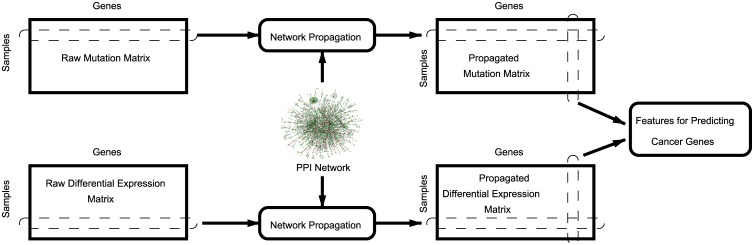
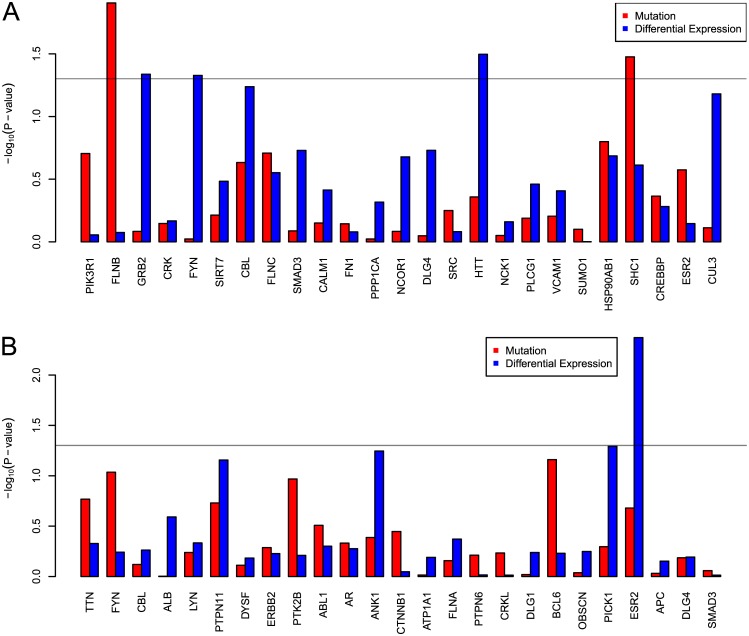
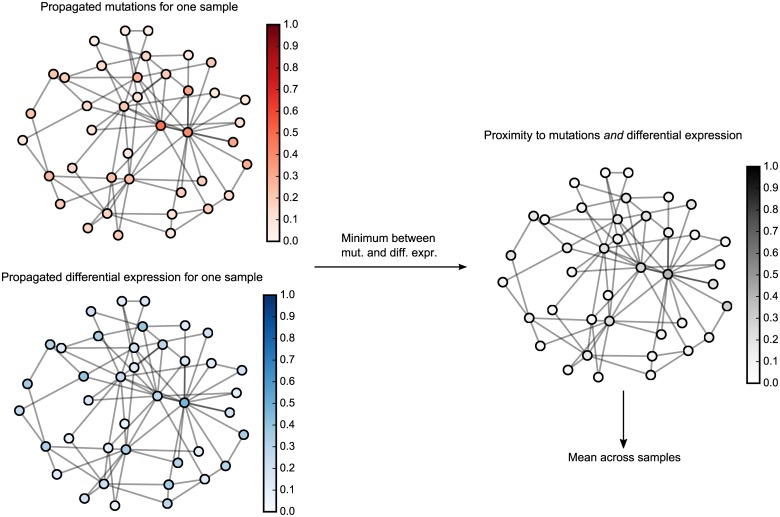
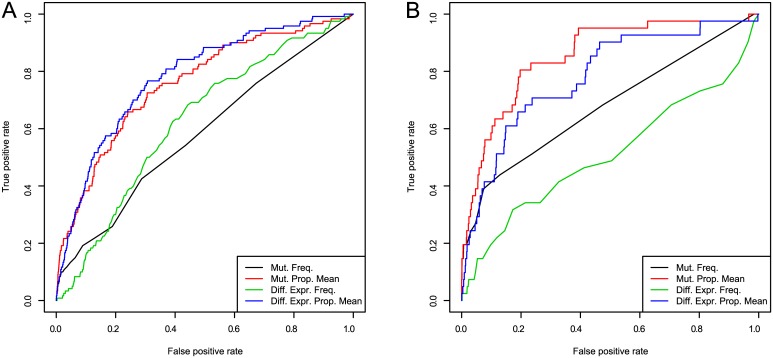
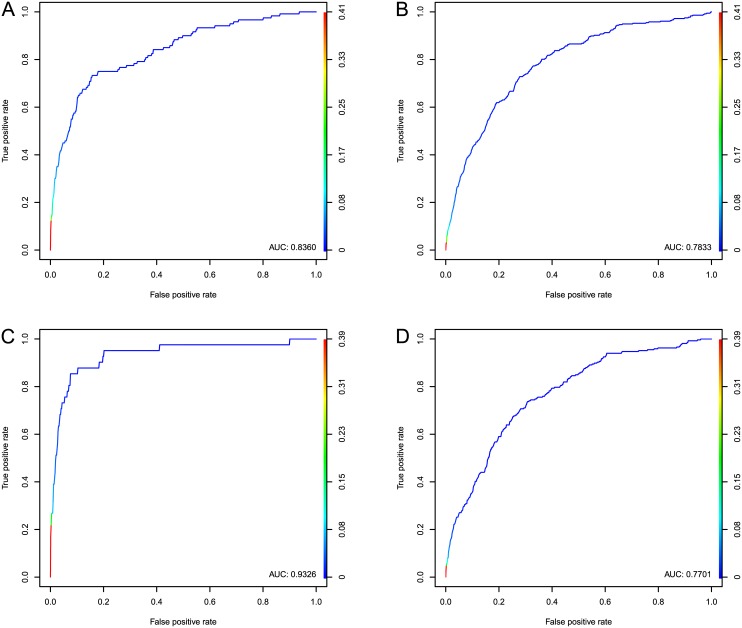
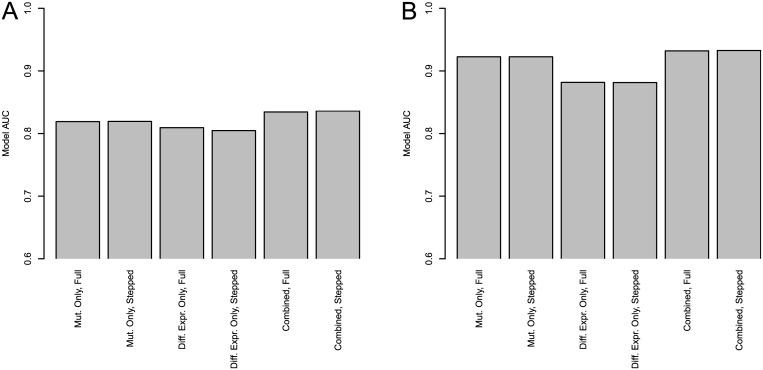
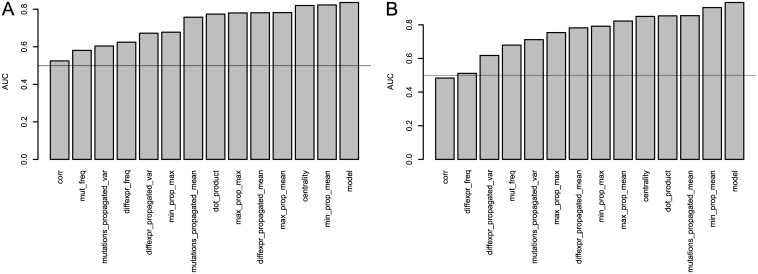
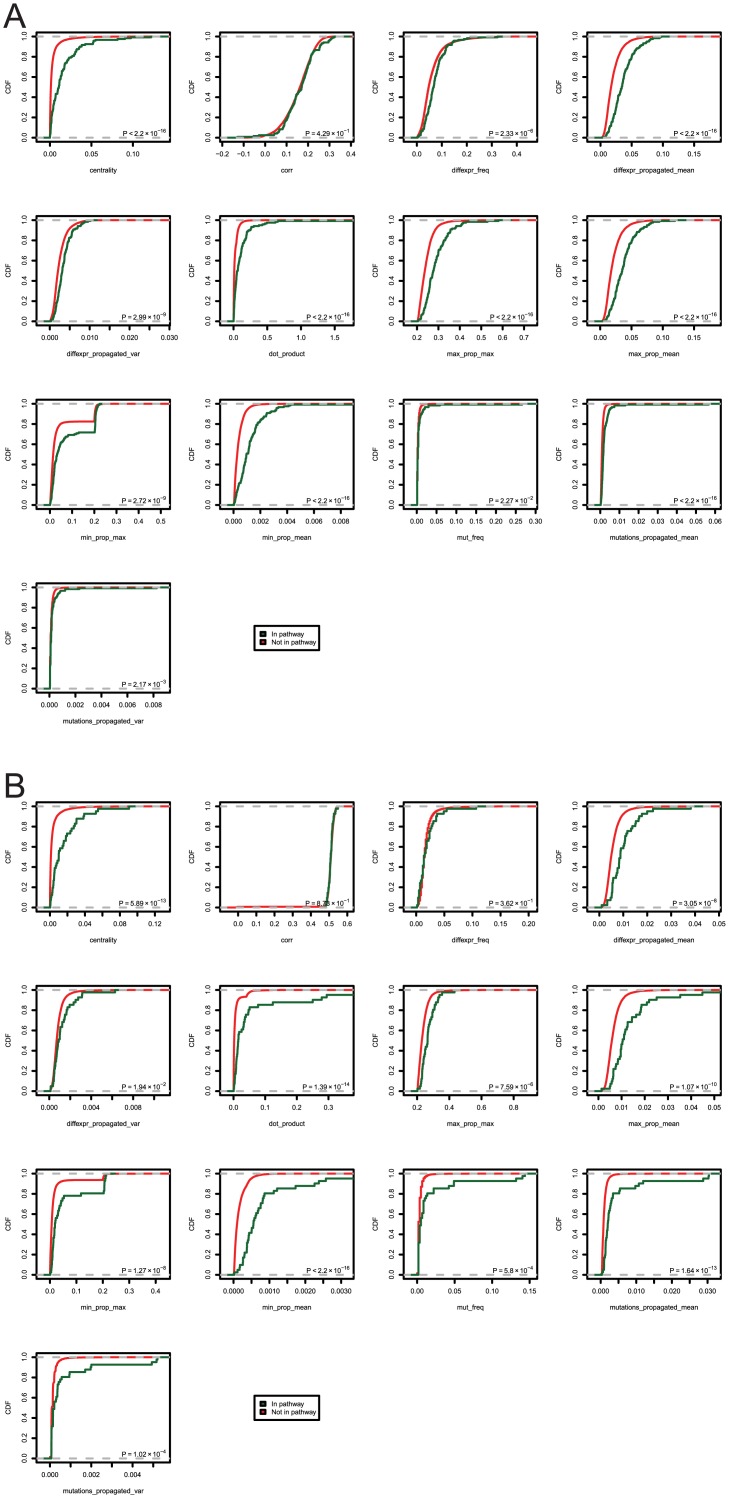
Development of high-throughput monitoring technologies enables interrogation of cancer samples at various levels of cellular activity. Capitalizing on these developments, various public efforts such as The Cancer Genome Atlas (TCGA) generate disparate omic data for large patient cohorts. As demonstrated by recent studies, these heterogeneous data sources provide the opportunity to gain insights into the molecular changes that drive cancer pathogenesis and progression. However, these insights are limited by the vast search space and as a result low statistical power to make new discoveries. In this paper, we propose methods for integrating disparate omic data using molecular interaction networks, with a view to gaining mechanistic insights into the relationship between molecular changes at different levels of cellular activity. Namely, we hypothesize that genes that play a role in cancer development and progression may be implicated by neither frequent mutation nor differential expression, and that network-based integration of mutation and differential expression data can reveal these "silent players". For this purpose, we utilize network-propagation algorithms to simulate the information flow in the cell at a sample-specific resolution. We then use the propagated mutation and expression signals to identify genes that are not necessarily mutated or differentially expressed genes, but have an essential role in tumor development and patient outcome. We test the proposed method on breast cancer and glioblastoma multiforme data obtained from TCGA. Our results show that the proposed method can identify important proteins that are not readily revealed by molecular data, providing insights beyond what can be gleaned by analyzing different types of molecular data in isolation.
高通量监测技术的发展使得能够在细胞活动的各个层面上对癌症样本进行检测。利用这些进展,诸如癌症基因组图谱(TCGA)之类的各种公共项目为大量患者队列生成了不同的组学数据。正如最近的研究所表明的,这些异质数据源提供了深入了解驱动癌症发病机制和进展的分子变化的机会。然而,这些见解受到巨大搜索空间的限制,因此发现新事物的统计能力较低。在本文中,我们提出了使用分子相互作用网络整合不同组学数据的方法,以期深入了解细胞活动不同层面上分子变化之间的关系。具体而言,我们假设在癌症发展和进展中起作用的基因可能既不会频繁发生突变,也不会出现差异表达,并且基于网络的突变和差异表达数据整合能够揭示这些“沉默参与者”。为此,我们利用网络传播算法以样本特异性分辨率模拟细胞中的信息流。然后,我们使用传播后的突变和表达信号来识别不一定是突变基因或差异表达基因,但在肿瘤发展和患者预后中起关键作用的基因。我们在从TCGA获得的乳腺癌和多形性胶质母细胞瘤数据上测试了所提出的方法。我们的结果表明,所提出的方法能够识别分子数据不易揭示的重要蛋白质,提供了超越单独分析不同类型分子数据所能获得的见解。