Sloan Max, Alegre-Abarrategui Javier, Potgieter Dawid, Kaufmann Anna-Kristin, Exley Richard, Deltheil Thierry, Threlfell Sarah, Connor-Robson Natalie, Brimblecombe Katherine, Wallings Rebecca, Cioroch Milena, Bannerman David M, Bolam J Paul, Magill Peter J, Cragg Stephanie J, Dodson Paul D, Wade-Martins Richard
Oxford Parkinson's Disease Centre, Department of Physiology, Anatomy and Genetics.
Oxford Parkinson's Disease Centre, Medical Research Council Brain Network Dynamics Unit, Department of Pharmacology and.
Hum Mol Genet. 2016 Mar 1;25(5):951-63. doi: 10.1093/hmg/ddv628. Epub 2016 Jan 6.
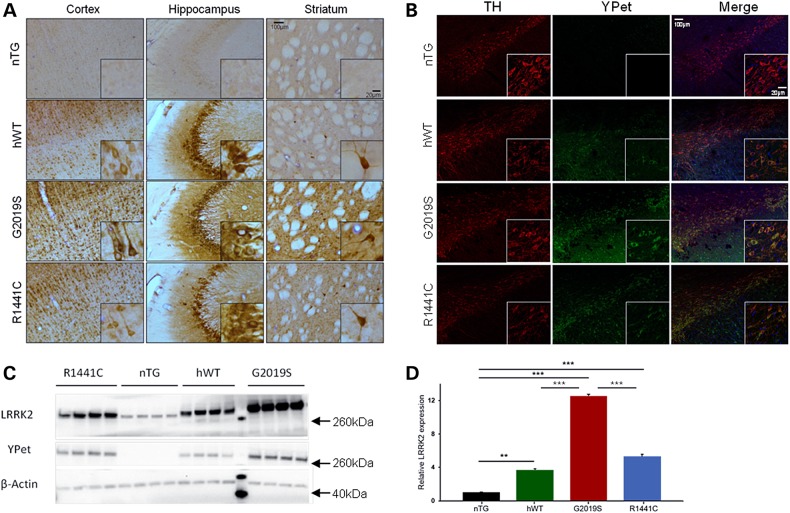
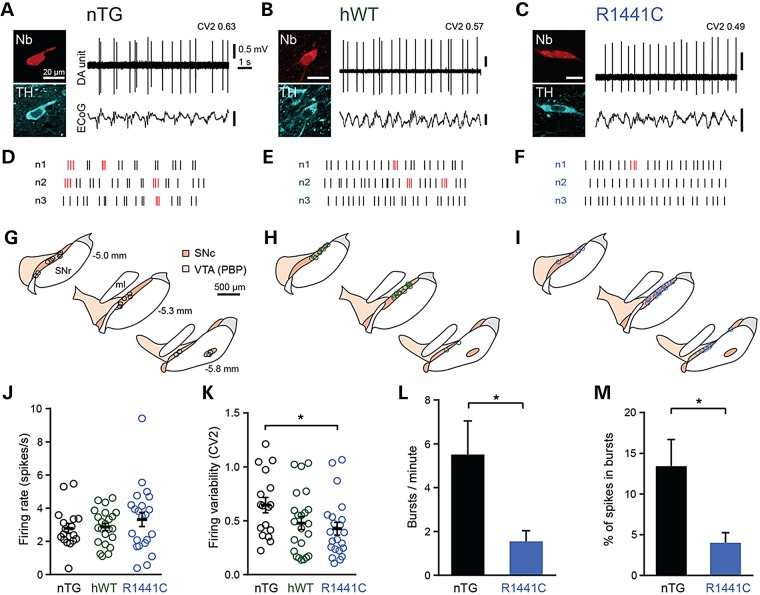
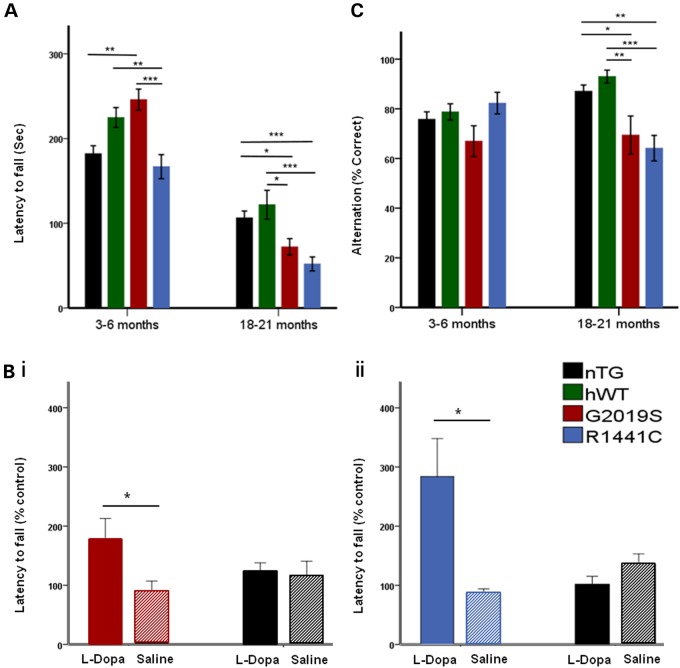
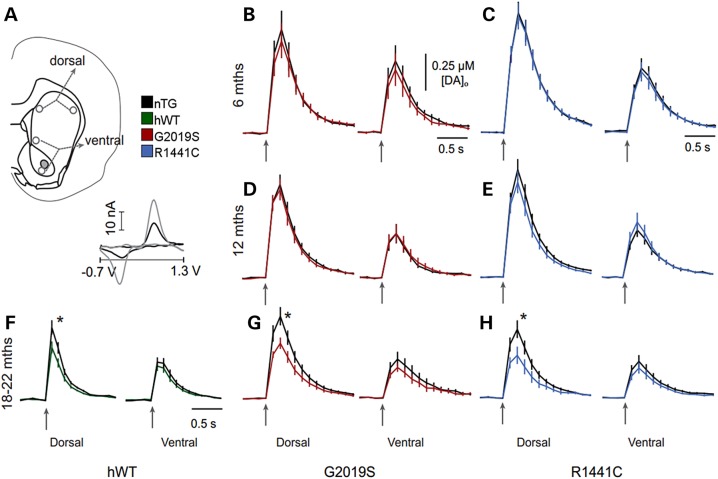
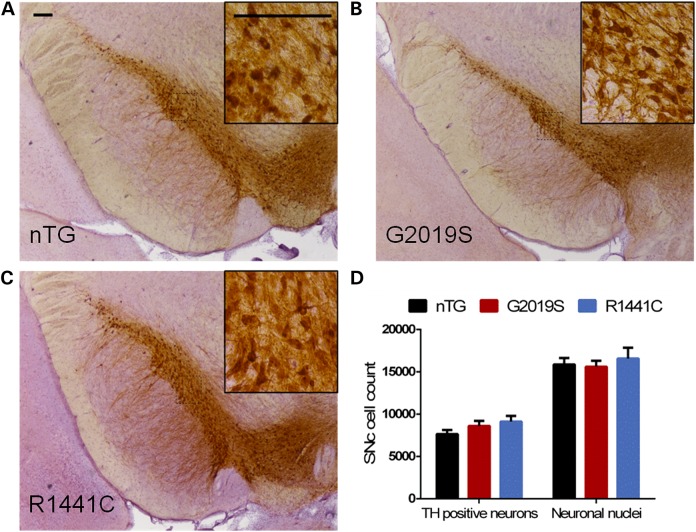
Mutations in leucine-rich repeat kinase 2 (LRRK2) lead to late-onset, autosomal dominant Parkinson's disease, characterized by the degeneration of dopamine neurons of the substantia nigra pars compacta, a deficit in dopamine neurotransmission and the development of motor and non-motor symptoms. The most prevalent Parkinson's disease LRRK2 mutations are located in the kinase (G2019S) and GTPase (R1441C) encoding domains of LRRK2. To better understand the sequence of events that lead to progressive neurophysiological deficits in vulnerable neurons and circuits in Parkinson's disease, we have generated LRRK2 bacterial artificial chromosome transgenic rats expressing either G2019S or R1441C mutant, or wild-type LRRK2, from the complete human LRRK2 genomic locus, including endogenous promoter and regulatory regions. Aged (18-21 months) G2019S and R1441C mutant transgenic rats exhibit L-DOPA-responsive motor dysfunction, impaired striatal dopamine release as determined by fast-scan cyclic voltammetry, and cognitive deficits. In addition, in vivo recordings of identified substantia nigra pars compacta dopamine neurons in R1441C LRRK2 transgenic rats reveal an age-dependent reduction in burst firing, which likely results in further reductions to striatal dopamine release. These alterations to dopamine circuit function occur in the absence of neurodegeneration or abnormal protein accumulation within the substantia nigra pars compacta, suggesting that nigrostriatal dopamine dysfunction precedes detectable protein aggregation and cell death in the development of Parkinson's disease. In conclusion, our longitudinal deep-phenotyping provides novel insights into how the genetic burden arising from human mutant LRRK2 manifests as early pathophysiological changes to dopamine circuit function and highlights a potential model for testing Parkinson's therapeutics.
富含亮氨酸重复激酶2(LRRK2)的突变会导致迟发性常染色体显性帕金森病,其特征是黑质致密部多巴胺神经元变性、多巴胺神经传递缺陷以及运动和非运动症状的出现。最常见的帕金森病LRRK2突变位于LRRK2的激酶(G2019S)和GTP酶(R1441C)编码结构域。为了更好地理解导致帕金森病中易损神经元和神经回路出现进行性神经生理缺陷的一系列事件,我们从完整的人类LRRK2基因组位点,包括内源性启动子和调控区域,生成了表达G2019S或R1441C突变体或野生型LRRK2的LRRK2细菌人工染色体转基因大鼠。老年(18 - 21个月)的G2019S和R1441C突变转基因大鼠表现出对左旋多巴有反应的运动功能障碍、通过快速扫描循环伏安法测定的纹状体多巴胺释放受损以及认知缺陷。此外,对R1441C LRRK2转基因大鼠中已鉴定的黑质致密部多巴胺神经元进行的体内记录显示,爆发性放电存在年龄依赖性减少,这可能导致纹状体多巴胺释放进一步减少。这些多巴胺回路功能的改变发生在黑质致密部没有神经变性或异常蛋白质积累的情况下,表明在帕金森病发展过程中,黑质纹状体多巴胺功能障碍先于可检测到的蛋白质聚集和细胞死亡。总之,我们的纵向深度表型分析为人类突变LRRK2产生的遗传负担如何表现为多巴胺回路功能的早期病理生理变化提供了新的见解,并突出了一个用于测试帕金森病治疗方法的潜在模型。