Department of Medical Sciences, Section of Pharmacology, and National Institute of Neuroscience, University of Ferrara, via Fossato di Mortara 17-19, 44121, Ferrara, Italy.
Department of Biology, University of Padova, Via Ugo Bassi 58/B, 35131, Padova, Italy.
Acta Neuropathol Commun. 2017 Mar 14;5(1):22. doi: 10.1186/s40478-017-0426-8.
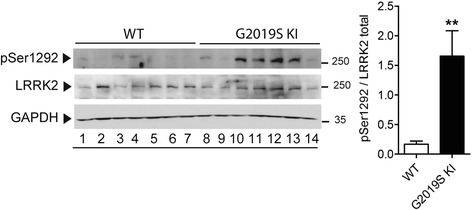
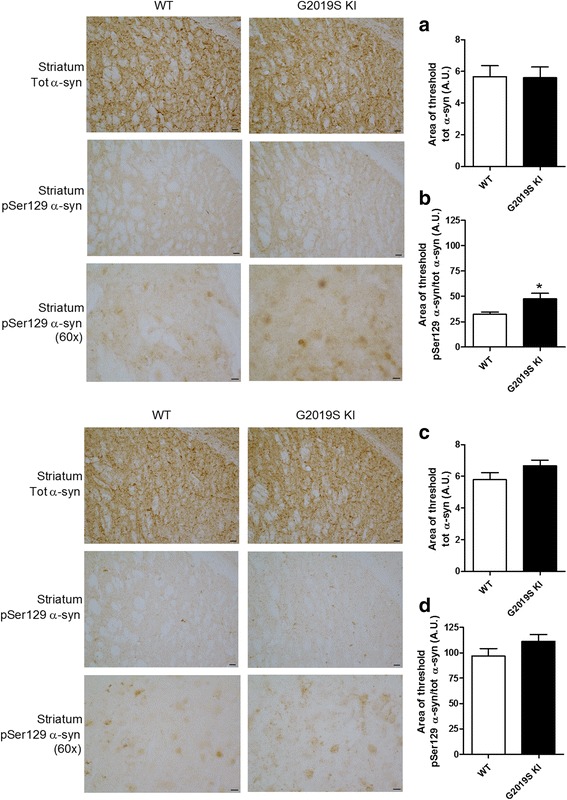
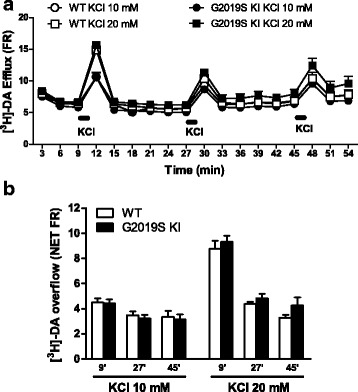
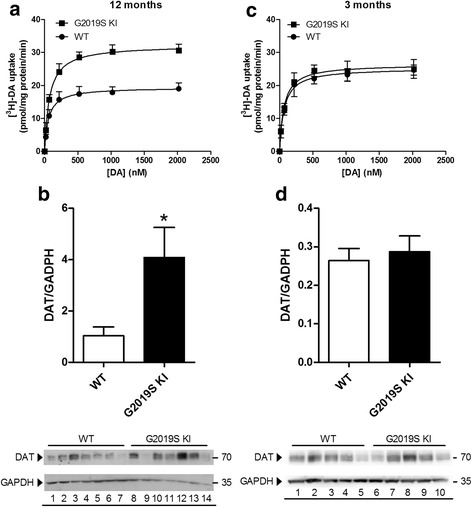
Mutations in the leucine-rich repeat kinase 2 (LRRK2) gene are the most common genetic cause of Parkinson's disease. Here, we investigated whether the G2019S LRRK2 mutation causes morphological and/or functional changes at nigro-striatal dopamine neurons. Density of striatal dopaminergic terminals, nigral cell counts, tyrosine hydroxylase protein levels as well as exocytotic dopamine release measured in striatal synaptosomes, or striatal extracellular dopamine levels monitored by in vivo microdialysis were similar between ≥12-month-old G2019S knock-in mice and wild-type controls. In vivo striatal dopamine release was insensitive to the LRRK2 inhibitor Nov-LRRK2-11, and was elevated by the membrane dopamine transporter blocker GBR-12783. However, G2019S knock-in mice showed a blunted neurochemical and motor activation response to GBR-12783 compared to wild-type controls. Western blot and dopamine uptake analysis revealed an increase in dopamine transporter levels and activity in the striatum of 12-month-old G2019S KI mice. This phenotype correlated with a reduction in vesicular monoamine transporter 2 levels and an enhancement of vesicular dopamine uptake, which was consistent with greater resistance to reserpine-induced hypolocomotion. These changes were not observed in 3-month-old mice. Finally, Western blot analysis revealed no genotype difference in striatal levels of endogenous α-synuclein or α-synuclein bound to DOPAL (a toxic metabolite of dopamine). However, Serine129-phosphorylated α-synuclein levels were higher in 12-month-old G2019S knock-in mice. Immunohistochemistry confirmed this finding, also showing no genotype difference in 3-month-old mice. We conclude that the G2019S mutation causes progressive dysfunctions of dopamine transporters, along with Serine129-phosphorylated α-synuclein overload, at striatal dopaminergic terminals, which are not associated with dopamine homeostasis dysregulation or neuron loss but might contribute to intrinsic dopaminergic terminal vulnerability. We propose G2019S knock-in mice as a presymptomatic Parkinson's disease model, useful to investigate the pathogenic interaction among genetics, aging, and internal or environmental factors leading to the disease.
LRRK2 基因突变是帕金森病最常见的遗传原因。在这里,我们研究了 G2019S LRRK2 突变是否会导致黑质纹状体多巴胺神经元的形态和/或功能改变。在 ≥12 个月大的 G2019S 敲入小鼠和野生型对照中,纹状体内多巴胺能末梢的密度、黑质细胞计数、酪氨酸羟化酶蛋白水平以及纹状体内突触小体中可测量的胞吐多巴胺释放或通过体内微透析监测的纹状体内细胞外多巴胺水平均相似。活体纹状体内多巴胺释放对 LRRK2 抑制剂 Nov-LRRK2-11 不敏感,并且被膜多巴胺转运体阻滞剂 GBR-12783 升高。然而,与野生型对照相比,G2019S 敲入小鼠对 GBR-12783 的神经化学和运动激活反应迟钝。Western blot 和多巴胺摄取分析显示,12 个月大的 G2019S KI 小鼠纹状体中的多巴胺转运体水平和活性增加。这种表型与囊泡单胺转运体 2 水平降低和囊泡多巴胺摄取增强相关,这与对利血平诱导的运动减少的抵抗力增强有关。这些变化在 3 个月大的小鼠中未观察到。最后,Western blot 分析显示,纹状体中内源性 α-突触核蛋白或与 DOPAL(多巴胺的有毒代谢物)结合的 α-突触核蛋白的基因型差异。然而,12 个月大的 G2019S 敲入小鼠中的 Serine129 磷酸化的 α-突触核蛋白水平更高。免疫组织化学证实了这一发现,在 3 个月大的小鼠中也没有基因型差异。我们得出结论,G2019S 突变导致纹状体多巴胺转运体的进行性功能障碍,以及 Serine129 磷酸化的 α-突触核蛋白过载,这与多巴胺稳态失调或神经元丢失无关,但可能有助于内在多巴胺能末梢的脆弱性。我们提出 G2019S 敲入小鼠作为帕金森病的前驱模型,可用于研究遗传、衰老以及导致疾病的内部或环境因素之间的致病相互作用。