Zhao Shanrong, Xi Li, Quan Jie, Xi Hualin, Zhang Ying, von Schack David, Vincent Michael, Zhang Baohong
PharmaTherapeutics Clinical R&D, Pfizer Worldwide Research and Development, Cambridge, MA, 02139, USA.
Computational Sciences Center of Emphasis, Pfizer Worldwide Research and Development, Cambridge, MA, 02139, USA.
BMC Genomics. 2016 Jan 8;17:39. doi: 10.1186/s12864-015-2356-9.
RNA sequencing (RNA-seq), a next-generation sequencing technique for transcriptome profiling, is being increasingly used, in part driven by the decreasing cost of sequencing. Nevertheless, the analysis of the massive amounts of data generated by large-scale RNA-seq remains a challenge. Multiple algorithms pertinent to basic analyses have been developed, and there is an increasing need to automate the use of these tools so as to obtain results in an efficient and user friendly manner. Increased automation and improved visualization of the results will help make the results and findings of the analyses readily available to experimental scientists.
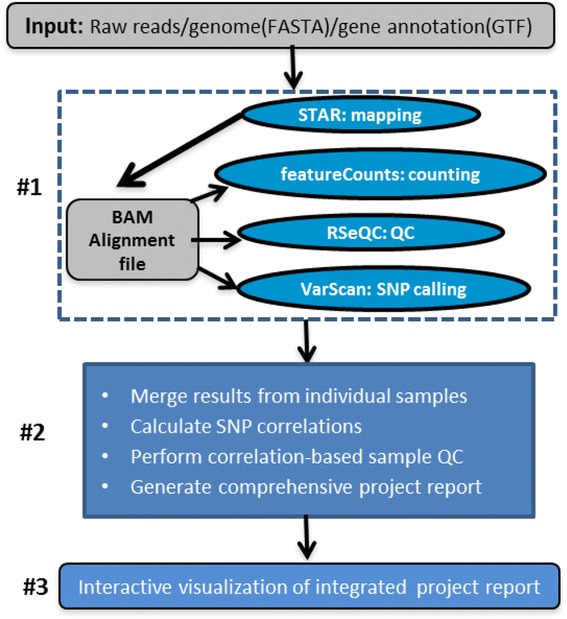
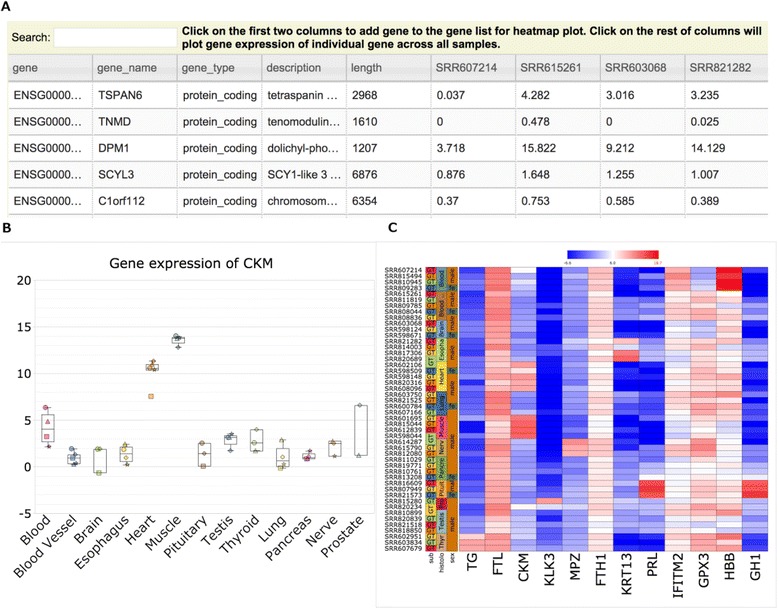
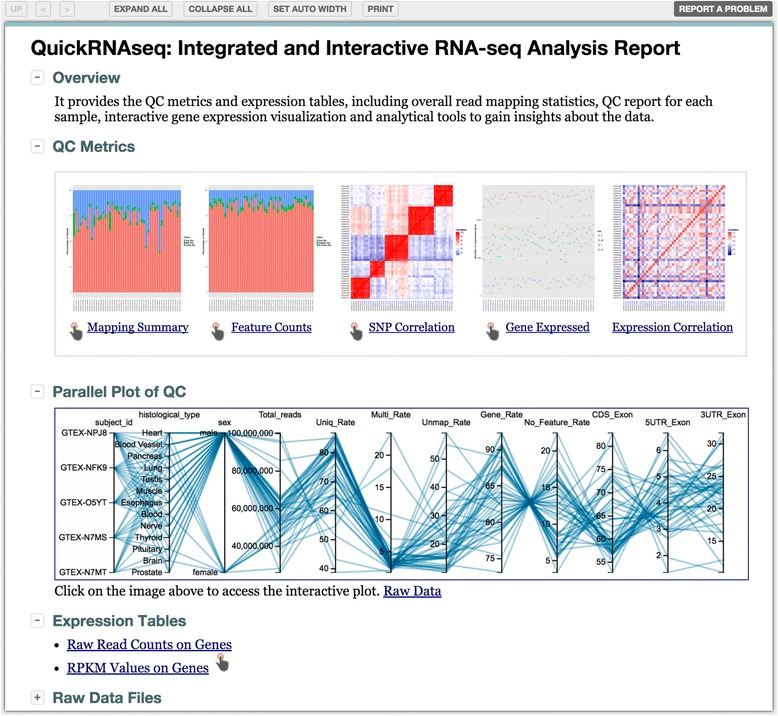
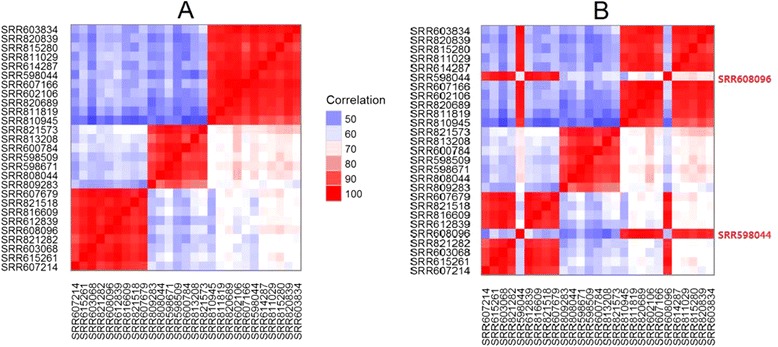
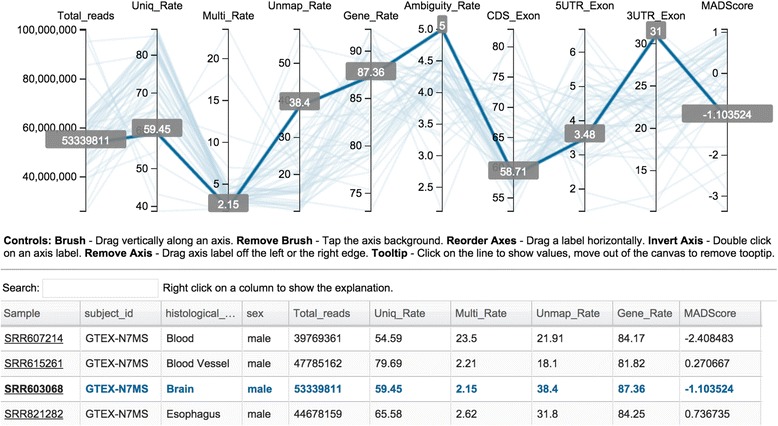
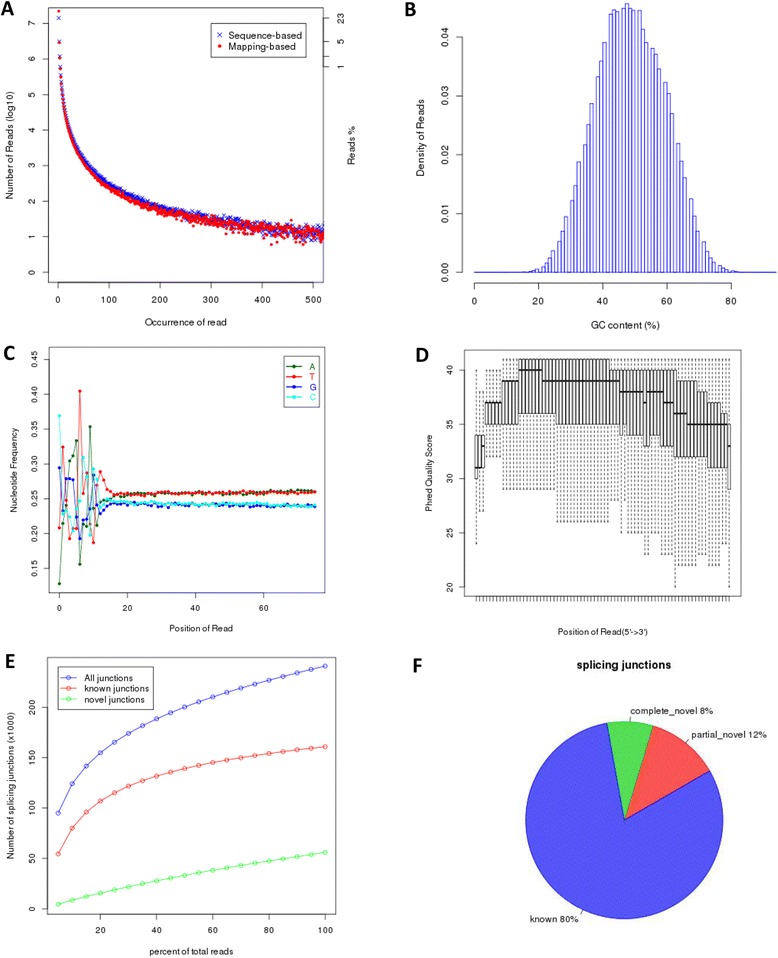
By combing the best open source tools developed for RNA-seq data analyses and the most advanced web 2.0 technologies, we have implemented QuickRNASeq, a pipeline for large-scale RNA-seq data analyses and visualization. The QuickRNASeq workflow consists of three main steps. In Step #1, each individual sample is processed, including mapping RNA-seq reads to a reference genome, counting the numbers of mapped reads, quality control of the aligned reads, and SNP (single nucleotide polymorphism) calling. Step #1 is computationally intensive, and can be processed in parallel. In Step #2, the results from individual samples are merged, and an integrated and interactive project report is generated. All analyses results in the report are accessible via a single HTML entry webpage. Step #3 is the data interpretation and presentation step. The rich visualization features implemented here allow end users to interactively explore the results of RNA-seq data analyses, and to gain more insights into RNA-seq datasets. In addition, we used a real world dataset to demonstrate the simplicity and efficiency of QuickRNASeq in RNA-seq data analyses and interactive visualizations. The seamless integration of automated capabilites with interactive visualizations in QuickRNASeq is not available in other published RNA-seq pipelines.
The high degree of automation and interactivity in QuickRNASeq leads to a substantial reduction in the time and effort required prior to further downstream analyses and interpretation of the analyses findings. QuickRNASeq advances primary RNA-seq data analyses to the next level of automation, and is mature for public release and adoption.
RNA测序(RNA-seq)是一种用于转录组分析的新一代测序技术,随着测序成本的降低,其应用越来越广泛。然而,对大规模RNA-seq产生的海量数据进行分析仍然是一项挑战。已经开发了多种与基础分析相关的算法,并且越来越需要自动化使用这些工具,以便以高效且用户友好的方式获得结果。提高自动化程度并改善结果的可视化将有助于使分析结果和发现易于被实验科学家获取。
通过结合为RNA-seq数据分析开发的最佳开源工具和最先进的Web 2.0技术,我们实现了QuickRNASeq,这是一个用于大规模RNA-seq数据分析和可视化的流程。QuickRNASeq工作流程包括三个主要步骤。在步骤1中,对每个单独的样本进行处理,包括将RNA-seq读数映射到参考基因组、计算映射读数的数量、对齐读数的质量控制以及单核苷酸多态性(SNP)检测。步骤1计算量很大,可以并行处理。在步骤2中,合并各个样本的结果,并生成一份综合且交互式的项目报告。报告中的所有分析结果都可以通过一个HTML入口网页访问。步骤3是数据解释和呈现步骤。这里实现的丰富可视化功能允许最终用户交互式地探索RNA-seq数据分析结果,并更深入地了解RNA-seq数据集。此外,我们使用一个实际数据集来证明QuickRNASeq在RNA-seq数据分析和交互式可视化方面的简单性和效率。QuickRNASeq中自动化功能与交互式可视化的无缝集成在其他已发表的RNA-seq流程中是不存在的。
QuickRNASeq的高度自动化和交互性使得在对分析结果进行进一步下游分析和解释之前所需的时间和精力大幅减少。QuickRNASeq将初级RNA-seq数据分析提升到了更高的自动化水平,并已成熟可供公众发布和采用。