Johnson Benjamin K, Scholz Matthew B, Teal Tracy K, Abramovitch Robert B
Department of Microbiology and Molecular Genetics, Michigan State University, East Lansing, MI, 48824, USA.
VANTAGE, Vanderbilt University, Nashville, TN, 37235, USA.
BMC Bioinformatics. 2016 Feb 4;17:66. doi: 10.1186/s12859-016-0923-y.
Many tools exist in the analysis of bacterial RNA sequencing (RNA-seq) transcriptional profiling experiments to identify differentially expressed genes between experimental conditions. Generally, the workflow includes quality control of reads, mapping to a reference, counting transcript abundance, and statistical tests for differentially expressed genes. In spite of the numerous tools developed for each component of an RNA-seq analysis workflow, easy-to-use bacterially oriented workflow applications to combine multiple tools and automate the process are lacking. With many tools to choose from for each step, the task of identifying a specific tool, adapting the input/output options to the specific use-case, and integrating the tools into a coherent analysis pipeline is not a trivial endeavor, particularly for microbiologists with limited bioinformatics experience.
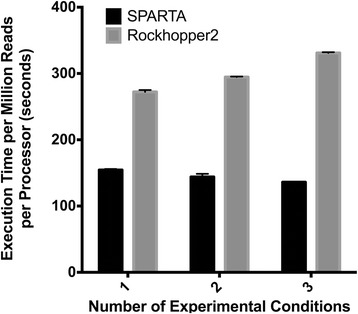
To make bacterial RNA-seq data analysis more accessible, we developed a Simple Program for Automated reference-based bacterial RNA-seq Transcriptome Analysis (SPARTA). SPARTA is a reference-based bacterial RNA-seq analysis workflow application for single-end Illumina reads. SPARTA is turnkey software that simplifies the process of analyzing RNA-seq data sets, making bacterial RNA-seq analysis a routine process that can be undertaken on a personal computer or in the classroom. The easy-to-install, complete workflow processes whole transcriptome shotgun sequencing data files by trimming reads and removing adapters, mapping reads to a reference, counting gene features, calculating differential gene expression, and, importantly, checking for potential batch effects within the data set. SPARTA outputs quality analysis reports, gene feature counts and differential gene expression tables and scatterplots.
SPARTA provides an easy-to-use bacterial RNA-seq transcriptional profiling workflow to identify differentially expressed genes between experimental conditions. This software will enable microbiologists with limited bioinformatics experience to analyze their data and integrate next generation sequencing (NGS) technologies into the classroom. The SPARTA software and tutorial are available at sparta.readthedocs.org.
在细菌RNA测序(RNA-seq)转录谱分析实验中,有许多工具可用于识别实验条件之间差异表达的基因。一般来说,工作流程包括读取数据的质量控制、映射到参考序列、计算转录本丰度以及对差异表达基因进行统计检验。尽管针对RNA-seq分析工作流程的每个组件都开发了众多工具,但缺乏易于使用的面向细菌的工作流程应用程序来组合多个工具并实现过程自动化。由于每个步骤都有许多工具可供选择,识别特定工具、使输入/输出选项适应特定用例以及将这些工具集成到连贯的分析流程中并非易事,特别是对于生物信息学经验有限的微生物学家而言。
为了使细菌RNA-seq数据分析更容易进行,我们开发了一个基于参考的细菌RNA-seq转录组自动分析简单程序(SPARTA)。SPARTA是一个用于单端Illumina读取的基于参考的细菌RNA-seq分析工作流程应用程序。SPARTA是一种交钥匙软件,简化了RNA-seq数据集的分析过程,使细菌RNA-seq分析成为可以在个人计算机上或课堂上进行的常规过程。这个易于安装的完整工作流程通过修剪读取数据和去除接头、将读取数据映射到参考序列、计算基因特征、计算差异基因表达,并且重要的是,检查数据集中的潜在批次效应,来处理全转录组鸟枪法测序数据文件。SPARTA输出质量分析报告、基因特征计数以及差异基因表达表和散点图。
SPARTA提供了一个易于使用的细菌RNA-seq转录谱工作流程,以识别实验条件之间差异表达的基因。该软件将使生物信息学经验有限的微生物学家能够分析他们的数据,并将下一代测序(NGS)技术引入课堂。SPARTA软件和教程可在sparta.readthedocs.org上获取。