Audoux Jérôme, Salson Mikaël, Grosset Christophe F, Beaumeunier Sacha, Holder Jean-Marc, Commes Thérèse, Philippe Nicolas
SeqOne, IRMB, CHRU de Montpellier -Hopital St Eloi, 80 avenue Augustin Fliche, Montpellier, 34295, France.
Institute of Computational Biology, Montpellier, 860, Rue Saint-Priest, Montpellier Cedex 5, 34095, France.
BMC Bioinformatics. 2017 Sep 29;18(1):428. doi: 10.1186/s12859-017-1831-5.
The evolution of next-generation sequencing (NGS) technologies has led to increased focus on RNA-Seq. Many bioinformatic tools have been developed for RNA-Seq analysis, each with unique performance characteristics and configuration parameters. Users face an increasingly complex task in understanding which bioinformatic tools are best for their specific needs and how they should be configured. In order to provide some answers to these questions, we investigate the performance of leading bioinformatic tools designed for RNA-Seq analysis and propose a methodology for systematic evaluation and comparison of performance to help users make well informed choices.
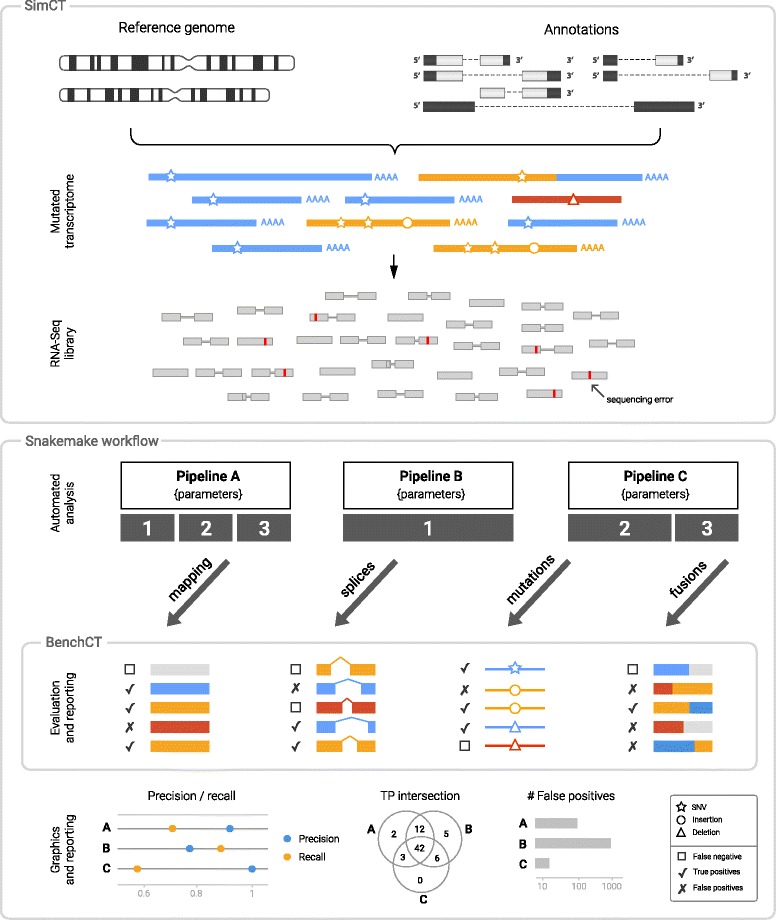
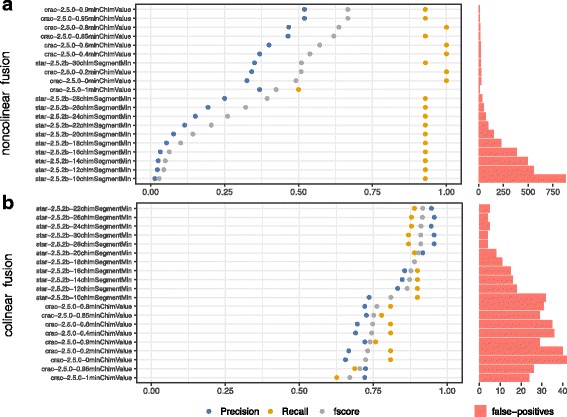
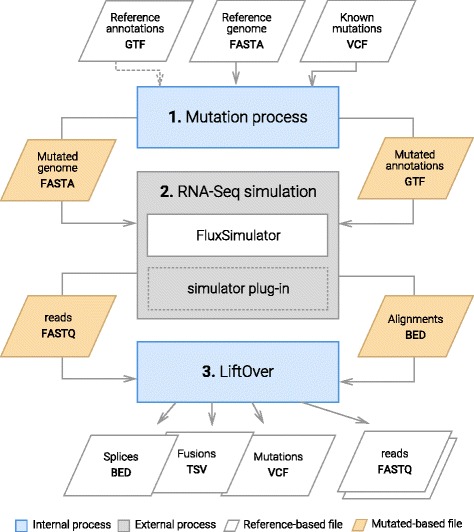
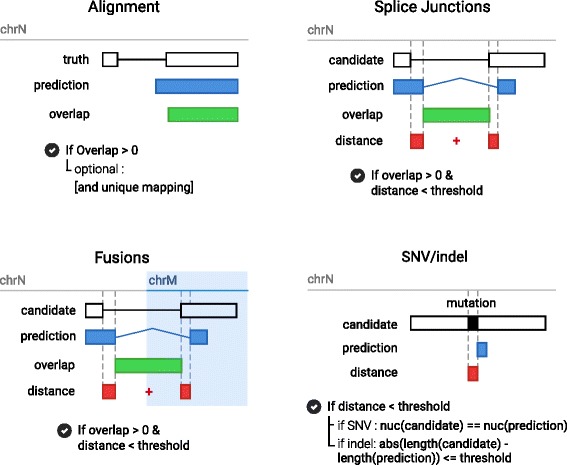
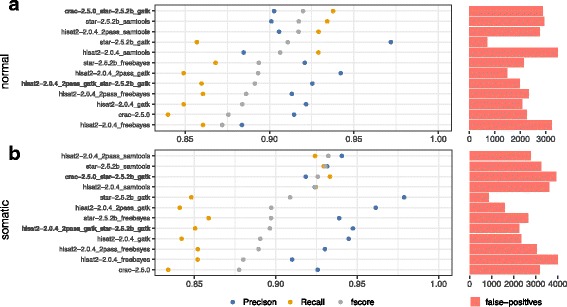
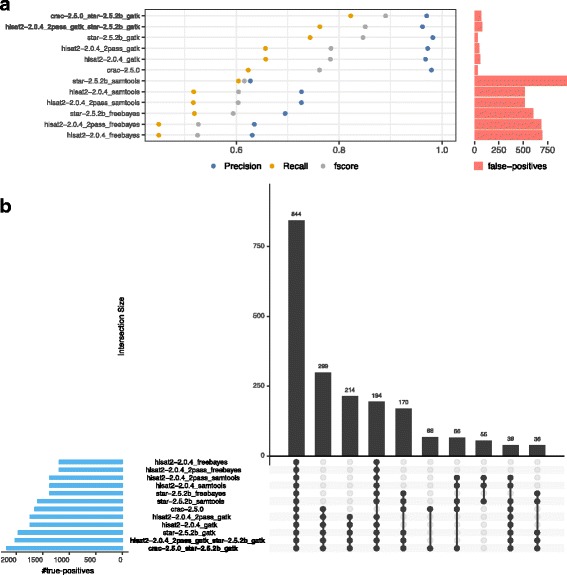
To evaluate RNA-Seq pipelines, we developed a suite of two benchmarking tools. SimCT generates simulated datasets that get as close as possible to specific real biological conditions accompanied by the list of genomic incidents and mutations that have been inserted. BenchCT then compares the output of any bioinformatics pipeline that has been run against a SimCT dataset with the simulated genomic and transcriptional variations it contains to give an accurate performance evaluation in addressing specific biological question. We used these tools to simulate a real-world genomic medicine question s involving the comparison of healthy and cancerous cells. Results revealed that performance in addressing a particular biological context varied significantly depending on the choice of tools and settings used. We also found that by combining the output of certain pipelines, substantial performance improvements could be achieved.
Our research emphasizes the importance of selecting and configuring bioinformatic tools for the specific biological question being investigated to obtain optimal results. Pipeline designers, developers and users should include benchmarking in the context of their biological question as part of their design and quality control process. Our SimBA suite of benchmarking tools provides a reliable basis for comparing the performance of RNA-Seq bioinformatics pipelines in addressing a specific biological question. We would like to see the creation of a reference corpus of data-sets that would allow accurate comparison between benchmarks performed by different groups and the publication of more benchmarks based on this public corpus. SimBA software and data-set are available at http://cractools.gforge.inria.fr/softwares/simba/ .
下一代测序(NGS)技术的发展使得人们对RNA测序(RNA-Seq)的关注日益增加。已经开发了许多用于RNA-Seq分析的生物信息学工具,每个工具都有独特的性能特征和配置参数。用户在理解哪些生物信息学工具最适合其特定需求以及应如何配置这些工具方面面临着日益复杂的任务。为了回答这些问题,我们研究了用于RNA-Seq分析的领先生物信息学工具的性能,并提出了一种系统评估和比较性能的方法,以帮助用户做出明智的选择。
为了评估RNA-Seq流程,我们开发了一套两个基准测试工具。SimCT生成模拟数据集,这些数据集尽可能接近特定的真实生物学条件,并附带已插入的基因组事件和突变列表。然后,BenchCT将针对SimCT数据集运行的任何生物信息学流程的输出与其中包含的模拟基因组和转录变异进行比较,以在解决特定生物学问题时给出准确的性能评估。我们使用这些工具模拟了一个涉及健康细胞和癌细胞比较的真实世界基因组医学问题。结果表明,根据所使用的工具和设置的选择,在解决特定生物学背景时的性能差异很大。我们还发现,通过组合某些流程的输出,可以实现显著的性能提升。
我们的研究强调了为所研究的特定生物学问题选择和配置生物信息学工具以获得最佳结果的重要性。流程设计者、开发者和用户应将针对其生物学问题的基准测试作为其设计和质量控制过程的一部分。我们的SimBA基准测试工具套件为比较RNA-Seq生物信息学流程在解决特定生物学问题时的性能提供了可靠的基础。我们希望看到创建一个数据集参考语料库,以便能够准确比较不同组执行的基准测试,并基于此公共语料库发布更多基准测试。SimBA软件和数据集可在http://cractools.gforge.inria.fr/softwares/simba/获取。