Wu Nicholas C, Du Yushen, Le Shuai, Young Arthur P, Zhang Tian-Hao, Wang Yuanyuan, Zhou Jian, Yoshizawa Janice M, Dong Ling, Li Xinmin, Wu Ting-Ting, Sun Ren
Department of Molecular and Medical Pharmacology, David Geffen School of Medicine, University of California, Los Angeles, 90095, CA, USA.
Molecular Biology InstituteUniversity of California, Los Angeles, 90095, CA, USA.
BMC Genomics. 2016 Jan 12;17:46. doi: 10.1186/s12864-015-2358-7.
Epistasis is one of the central themes in viral evolution due to its importance in drug resistance, immune escape, and interspecies transmission. However, there is a lack of experimental approach to systematically probe for epistatic residues.
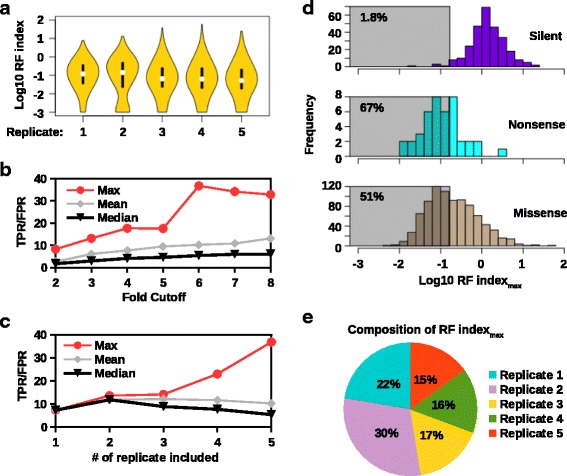
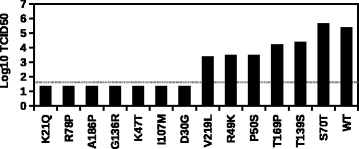
By utilizing the information from natural occurring sequences and high-throughput genetics, this study established a novel strategy to identify epistatic residues. The rationale is that a substitution that is deleterious in one strain may be prevalent in nature due to the presence of a naturally occurring compensatory substitution. Here, high-throughput genetics was applied to influenza A virus M segment to systematically identify deleterious substitutions. Comparison with natural sequence variation showed that a deleterious substitution M1 Q214H was prevalent in circulating strains. A coevolution analysis was then performed and indicated that M1 residues 121, 207, 209, and 214 naturally coevolved as a group. Subsequently, we experimentally validated that M1 A209T was a compensatory substitution for M1 Q214H.
This work provided a proof-of-concept to identify epistatic residues by coupling high-throughput genetics with phylogenetic information. In particular, we were able to identify an epistatic interaction between M1 substitutions A209T and Q214H. This analytic strategy can potentially be adapted to study any protein of interest, provided that the information on natural sequence variants is available.
上位性是病毒进化的核心主题之一,因为它在耐药性、免疫逃逸和种间传播中具有重要意义。然而,目前缺乏系统探测上位性残基的实验方法。
本研究利用天然序列信息和高通量遗传学,建立了一种识别上位性残基的新策略。其基本原理是,一个在某一毒株中有害的替换,可能由于存在一个天然的补偿性替换而在自然界中普遍存在。在此,高通量遗传学被应用于甲型流感病毒M基因片段,以系统地识别有害替换。与天然序列变异的比较表明,有害替换M1 Q214H在流行毒株中普遍存在。随后进行的共进化分析表明,M1蛋白的121、207、209和214位残基自然地作为一个群体共同进化。随后,我们通过实验验证了M1 A209T是M1 Q214H的补偿性替换。
这项工作通过将高通量遗传学与系统发育信息相结合,为识别上位性残基提供了概念验证。特别是,我们能够识别M1替换A209T和Q214H之间的上位性相互作用。只要有天然序列变异的信息,这种分析策略有可能适用于研究任何感兴趣的蛋白质。