Riaz Nasir, Leung Preston, Barton Kirston, Smith Martin A, Carswell Shaun, Bull Rowena, Lloyd Andrew R, Rodrigo Chaturaka
Kirby Institute, UNSW Sydney, Sydney, NSW, 2052, Australia.
Department of Microbiology, Hazara University, KPK, Maneshra, 21120, Pakistan.
BMC Genomics. 2021 Mar 2;22(1):148. doi: 10.1186/s12864-021-07460-1.
Hepatitis C (HCV) and many other RNA viruses exist as rapidly mutating quasi-species populations in a single infected host. High throughput characterization of full genome, within-host variants is still not possible despite advances in next generation sequencing. This limitation constrains viral genomic studies that depend on accurate identification of hemi-genome or whole genome, within-host variants, especially those occurring at low frequencies. With the advent of third generation long read sequencing technologies, including Oxford Nanopore Technology (ONT) and PacBio platforms, this problem is potentially surmountable. ONT is particularly attractive in this regard due to the portable nature of the MinION sequencer, which makes real-time sequencing in remote and resource-limited locations possible. However, this technology (termed here 'nanopore sequencing') has a comparatively high technical error rate. The present study aimed to assess the utility, accuracy and cost-effectiveness of nanopore sequencing for HCV genomes. We also introduce a new bioinformatics tool (Nano-Q) to differentiate within-host variants from nanopore sequencing.
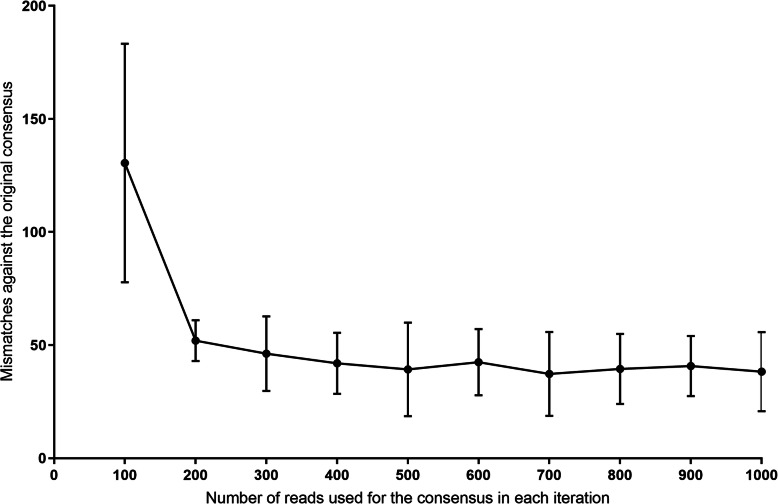
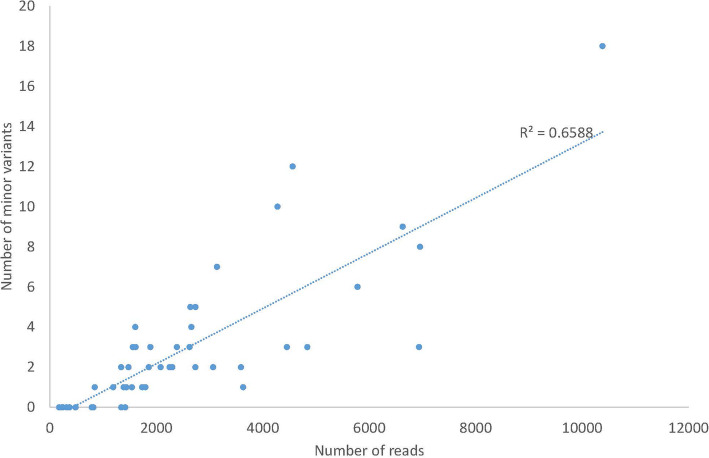
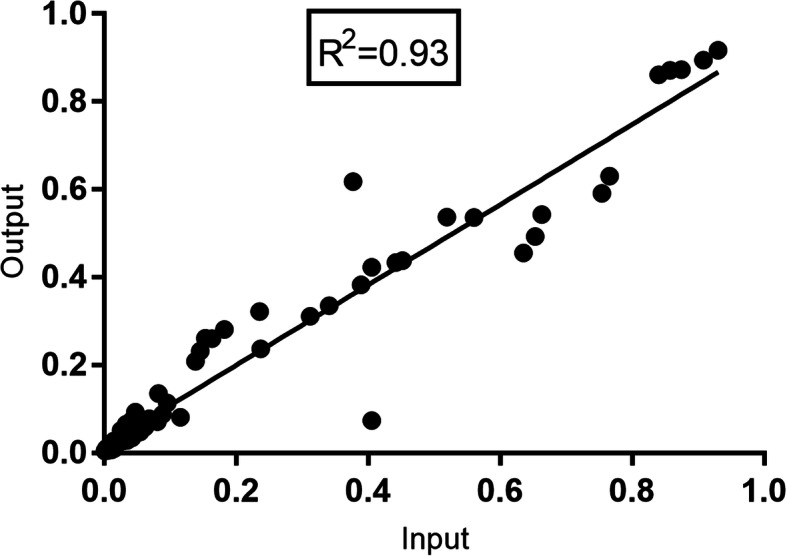
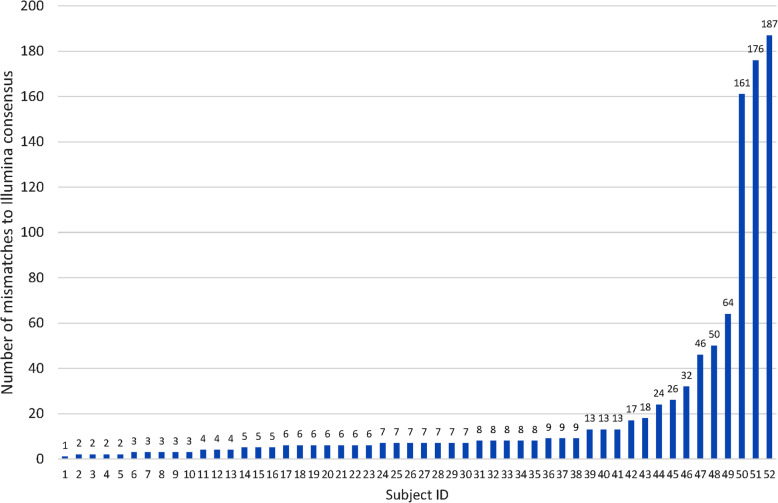
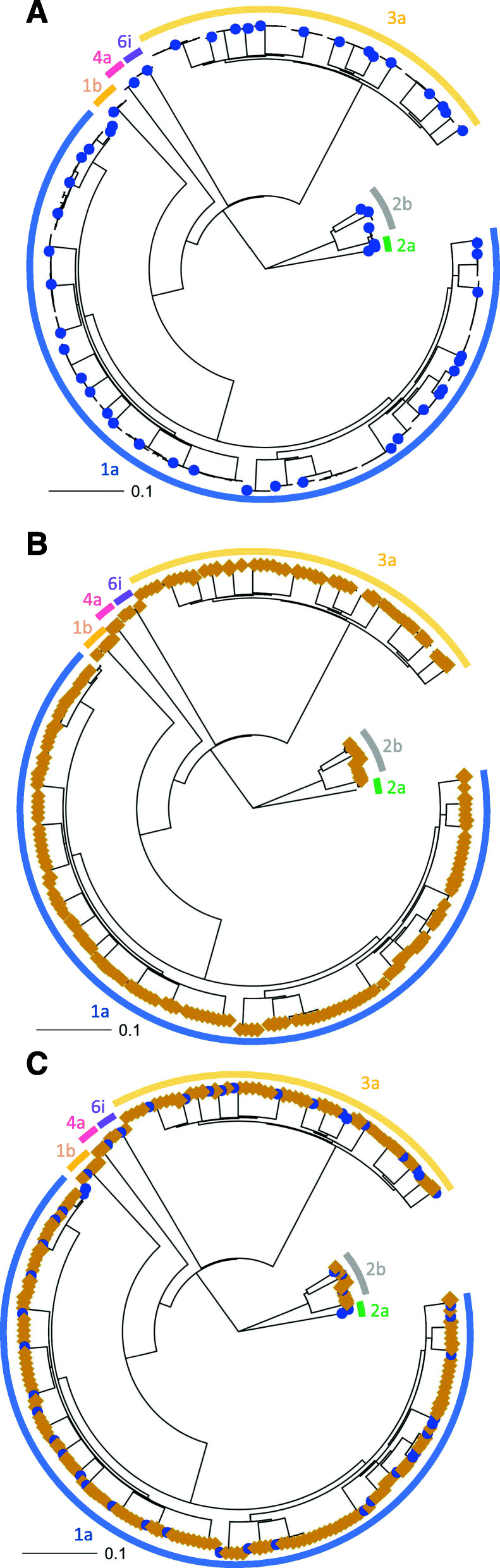
The Nanopore platform, when the coverage exceeded 300 reads, generated comparable consensus sequences to Illumina sequencing. Using HCV Envelope plasmids (~ 1800 nt) mixed in known proportions, the capacity of nanopore sequencing to reliably identify variants with an abundance as low as 0.1% was demonstrated, provided the autologous reference sequence was available to identify the matching reads. Successful pooling and nanopore sequencing of 52 samples from patients with HCV infection demonstrated its cost effectiveness (AUD$ 43 per sample with nanopore sequencing versus $100 with paired-end short read technology). The Nano-Q tool successfully separated between-host sequences, including those from the same subtype, by bulk sorting and phylogenetic clustering without an autologous reference sequence (using only a subtype-specific generic reference). The pipeline also identified within-host viral variants and their abundance when the parameters were appropriately adjusted.
Cost effective HCV whole genome sequencing and within-host variant identification without haplotype reconstruction are potential advantages of nanopore sequencing.
丙型肝炎病毒(HCV)和许多其他RNA病毒在单个受感染宿主中以快速变异的准种群体形式存在。尽管下一代测序技术取得了进展,但仍无法对全基因组、宿主内变异体进行高通量表征。这一限制制约了依赖于准确识别半基因组或全基因组、宿主内变异体(尤其是低频出现的变异体)的病毒基因组研究。随着包括牛津纳米孔技术(ONT)和PacBio平台在内的第三代长读长测序技术的出现,这个问题有可能得到解决。ONT在这方面特别有吸引力,因为MinION测序仪具有便携性,使得在偏远和资源有限的地点进行实时测序成为可能。然而,这项技术(这里称为“纳米孔测序”)具有相对较高的技术错误率。本研究旨在评估纳米孔测序用于HCV基因组的实用性、准确性和成本效益。我们还引入了一种新的生物信息学工具(Nano-Q)来区分纳米孔测序中的宿主内变异体。
当覆盖度超过300次读取时,纳米孔平台生成的一致性序列与Illumina测序相当。使用以已知比例混合的HCV包膜质粒(约1800 nt),证明了纳米孔测序能够可靠地识别丰度低至0.1%的变异体,前提是有自体参考序列来识别匹配的读取。对52例HCV感染患者的样本进行成功混合和纳米孔测序,证明了其成本效益(纳米孔测序每个样本43澳元,而双端短读长技术为100澳元)。Nano-Q工具通过批量分类和系统发育聚类成功分离了宿主间序列,包括来自同一亚型的序列,无需自体参考序列(仅使用亚型特异性通用参考)。当参数适当调整时,该流程还能识别宿主内病毒变异体及其丰度。
具有成本效益的HCV全基因组测序以及无需单倍型重建的宿主内变异体识别是纳米孔测序的潜在优势。