Uddin Reaz, Sufian Muhammad
Dr. Panjwani Center for Molecular Medicine and Drug Research, International Center for Chemical and Biological Sciences, University of Karachi, Karachi 75270, Pakistan.
Prince of Wales Clinical School, Faculty of Medicine, UNSW Australia, Sydney, Australia.
PLoS One. 2016 Jan 22;11(1):e0146796. doi: 10.1371/journal.pone.0146796. eCollection 2016.
Infections caused by Salmonella enterica, a Gram-negative facultative anaerobic bacteria belonging to the family of Enterobacteriaceae, are major threats to the health of humans and animals. The recent availability of complete genome data of pathogenic strains of the S. enterica gives new avenues for the identification of drug targets and drug candidates. We have used the genomic and metabolic pathway data to identify pathways and proteins essential to the pathogen and absent from the host.
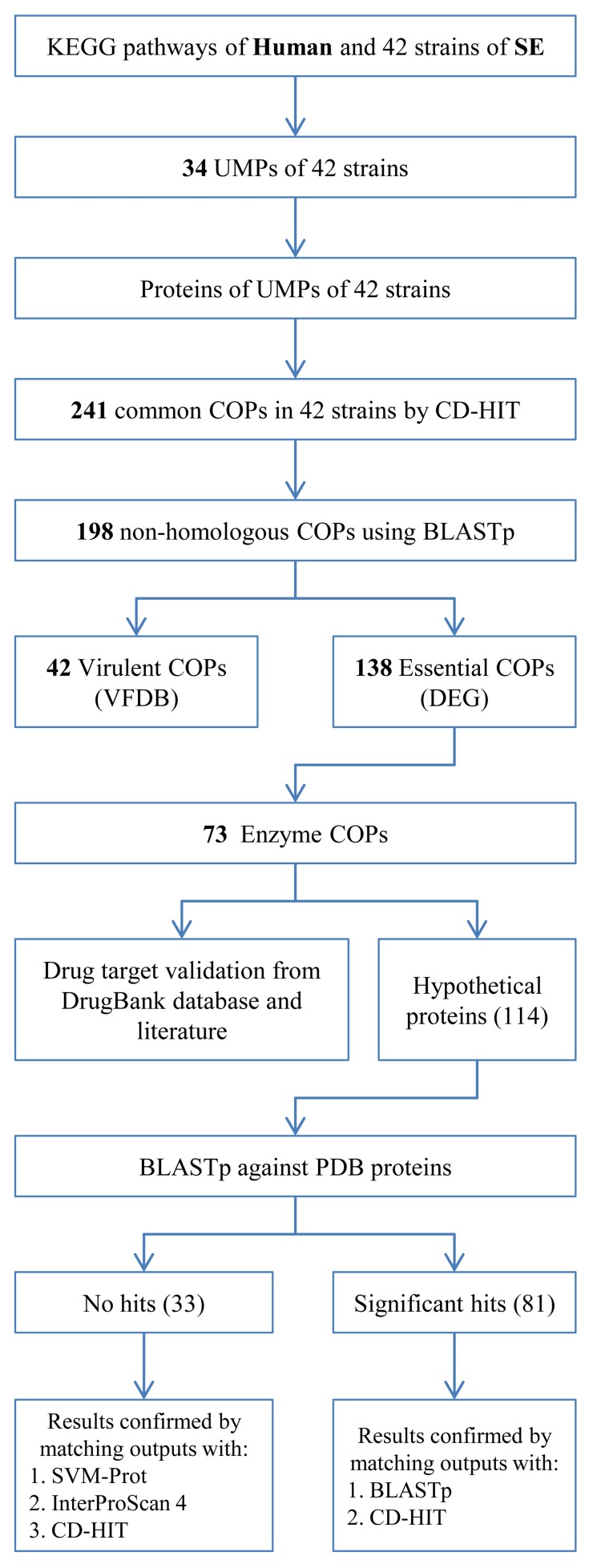
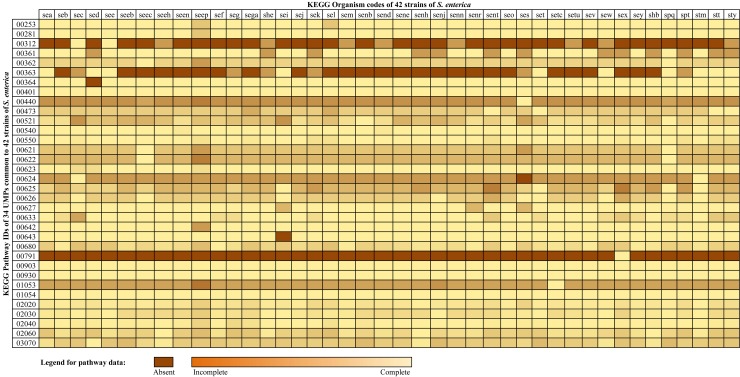
We took the whole proteome sequence data of 42 strains of S. enterica and Homo sapiens along with KEGG-annotated metabolic pathway data, clustered proteins sequences using CD-HIT, identified essential genes using DEG database and discarded S. enterica homologs of human proteins in unique metabolic pathways (UMPs) and characterized hypothetical proteins with SVM-prot and InterProScan. Through this core proteomic analysis we have identified enzymes essential to the pathogen.
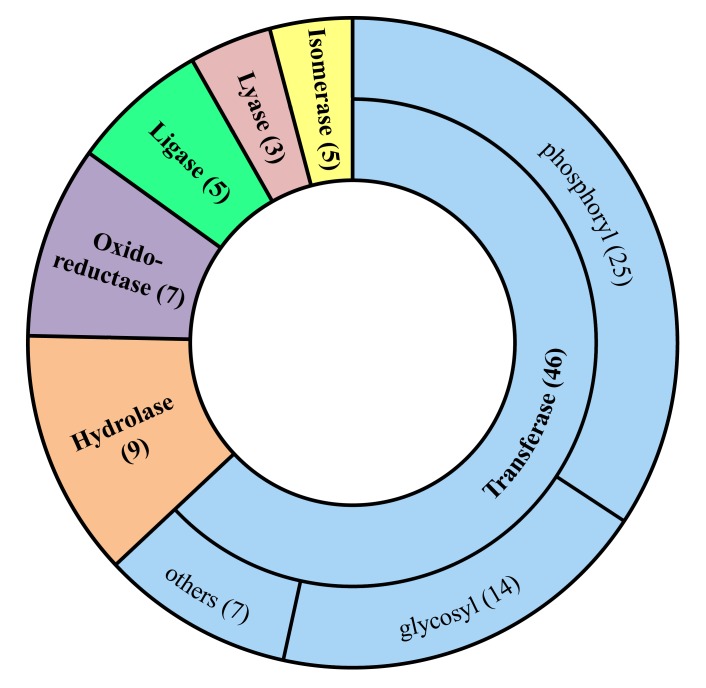
The identification of 73 enzymes common in 42 strains of S. enterica is the real strength of the current study. We proposed all 73 unexplored enzymes as potential drug targets against the infections caused by the S. enterica. The study is comprehensive around S. enterica and simultaneously considered every possible pathogenic strain of S. enterica. This comprehensiveness turned the current study significant since, to the best of our knowledge it is the first subtractive core proteomic analysis of the unique metabolic pathways applied to any pathogen for the identification of drug targets. We applied extensive computational methods to shortlist few potential drug targets considering the druggability criteria e.g. Non-homologous to the human host, essential to the pathogen and playing significant role in essential metabolic pathways of the pathogen (i.e. S. enterica). In the current study, the subtractive proteomics through a novel approach was applied i.e. by considering only proteins of the unique metabolic pathways of the pathogens and mining the proteomic data of all completely sequenced strains of the pathogen, thus improving the quality and application of the results. We believe that the sharing of the knowledge from this study would eventually lead to bring about novel and unique therapeutic regimens against the infections caused by the S. enterica.
肠炎沙门氏菌是一种革兰氏阴性兼性厌氧菌,属于肠杆菌科,其引起的感染对人类和动物的健康构成重大威胁。肠炎沙门氏菌致病菌株完整基因组数据的近期可得为药物靶点和候选药物的鉴定提供了新途径。我们利用基因组和代谢途径数据来鉴定病原体所必需且宿主中不存在的途径和蛋白质。
我们获取了42株肠炎沙门氏菌和智人的全蛋白质组序列数据以及KEGG注释的代谢途径数据,使用CD-HIT对蛋白质序列进行聚类,利用DEG数据库鉴定必需基因,并在独特代谢途径(UMPs)中剔除人类蛋白质的肠炎沙门氏菌同源物,并用SVM-prot和InterProScan对假设蛋白质进行表征。通过这种核心蛋白质组学分析,我们鉴定出了病原体所必需的酶。
鉴定出42株肠炎沙门氏菌中常见的73种酶是本研究的真正优势所在。我们提出将所有73种未被探索的酶作为针对肠炎沙门氏菌引起感染的潜在药物靶点。该研究围绕肠炎沙门氏菌展开且全面,同时考虑了肠炎沙门氏菌的每一种可能致病菌株。这种全面性使本研究具有重要意义,因为据我们所知,这是首次将独特代谢途径的减法核心蛋白质组学分析应用于任何病原体以鉴定药物靶点。我们应用了广泛的计算方法,根据可成药标准(如与人类宿主非同源、对病原体必需且在病原体(即肠炎沙门氏菌)的必需代谢途径中起重要作用)筛选出少数潜在药物靶点。在本研究中,采用了一种新颖的方法进行减法蛋白质组学研究,即仅考虑病原体独特代谢途径中的蛋白质,并挖掘该病原体所有完全测序菌株的蛋白质组数据,从而提高了结果的质量和应用价值。我们相信,分享本研究的知识最终将带来针对肠炎沙门氏菌引起感染的新颖独特治疗方案。