Wang Guoliang, Fan Rui, Ji Ruirui, Zou Wenxin, Penny Daniel J, Varghese Nidhy P, Fan Yuxin
John Welsh Cardiovascular Diagnostic Laboratory, Section of Cardiology, Department of Pediatrics, Texas Children's Hospital, Baylor College of Medicine, 1102 Bates Ave, Suite 430.09, Houston, TX, 77030, USA.
Department of Pediatrics, Xijing Hospital, The Fourth Military Medical University, Xi'an, 710032, Shanxi, China.
BMC Pulm Med. 2016 Jan 22;16:17. doi: 10.1186/s12890-016-0183-7.
Pulmonary arterial hypertension (PAH) is a rare, progressive, fatal vascular disorder. Genetic predisposition plays vital roles in the development of PAH, with most mutations being identified in genes involved in the transforming growth factor beta (TGF-β) signaling pathways. Defects in the BMP9 gene have been documented in hereditary hemorrhagic telangiectasia (HHT), the most common inherited vascular disorder, which is occasionally associated with PAH. Selective enhancement of endothelial BMPR2 with BMP9 reverses pulmonary arterial hypertension.
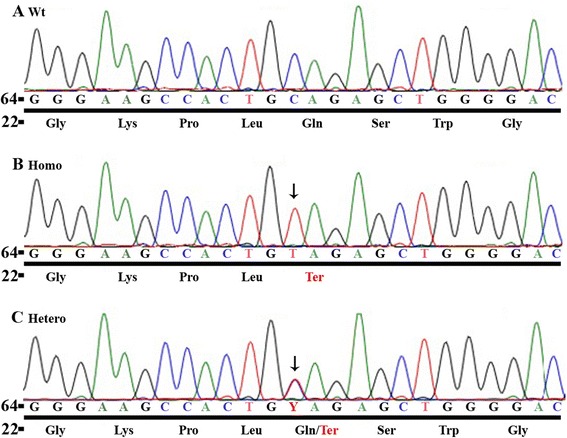
We report the case of a 5-year-old Hispanic boy who was diagnosed with severe PAH and right heart failure at 3 years of age. During his stay in the pediatric intensive care unit, treatment was initiated with inhaled nitric oxide and intravenous epoprostenol; he subsequently was transitioned to treprostinil, sildenafil, and prophylactic enoxaparin. Now, two years later, the child is asymptomatic on sildenafil, bosentan, subcutaneous treprostinil, and warfarin. Genetic screening revealed a novel homozygous nonsense mutation in the BMP9 gene (c.76C > T; p.Gln26Ter). The child had no telangiectasias or arteriovenous malformations; family history also was negative. Subsequent parental testing showed both parents were heterozygous for the same mutation, indicating that the child inherited the BMP9 mutant allele from each parent.
To our knowledge, this is the first report of a BMP9 mutation in a patient with PAH. The homozygous nonsense mutation may account for the early onset and severity of PAH in this patient and also fit the 'two-hit' model we proposed previously. The absence of clinical symptoms for PAH in the parents may be due to incomplete penetrance or various expressivities of the BMP9 mutations. Our study expands the spectrum of phenotypes related to BMP9 mutations.
肺动脉高压(PAH)是一种罕见的、进行性的、致命的血管疾病。遗传易感性在PAH的发生发展中起着至关重要的作用,大多数突变存在于参与转化生长因子β(TGF-β)信号通路的基因中。BMP9基因缺陷已在遗传性出血性毛细血管扩张症(HHT)中得到证实,HHT是最常见的遗传性血管疾病,偶尔与PAH相关。用BMP9选择性增强内皮BMPR2可逆转肺动脉高压。
我们报告了一名5岁西班牙裔男孩的病例,他在3岁时被诊断为重度PAH和右心衰竭。在儿科重症监护病房住院期间,开始使用吸入一氧化氮和静脉注射依前列醇进行治疗;随后他改用曲前列尼尔、西地那非和预防性依诺肝素。两年后的现在,该患儿在服用西地那非、波生坦、皮下注射曲前列尼尔和华法林时无症状。基因筛查发现BMP9基因存在一种新的纯合无义突变(c.76C>T;p.Gln26Ter)。该患儿无毛细血管扩张或动静脉畸形;家族史也为阴性。随后对其父母的检测显示,父母双方均为该突变的杂合子,这表明该患儿从父母双方各继承了一个BMP9突变等位基因。
据我们所知,这是PAH患者中BMP9突变的首例报告。该纯合无义突变可能是该患者PAH早发和严重程度的原因,也符合我们之前提出的“双打击”模型。父母中PAH无临床症状可能是由于BMP9突变的不完全外显或不同的表现度。我们的研究扩展了与BMP9突变相关的表型谱。