Xavier A, Muir William M, Rainey Katy M
Department of Agronomy, Purdue University, Lilly Hall of Life Sciences, 915 W. State St., West Lafayette, Indiana, 47907, USA.
Department of Animal Science, Purdue University, Lilly Hall of Life Sciences, 915 W. State St., West Lafayette, Indiana, 47907, USA.
BMC Bioinformatics. 2016 Feb 2;17:55. doi: 10.1186/s12859-016-0899-7.
Success in genome-wide association studies and marker-assisted selection depends on good phenotypic and genotypic data. The more complete this data is, the more powerful will be the results of analysis. Nevertheless, there are next-generation technologies that seek to provide genotypic information in spite of great proportions of missing data. The procedures these technologies use to impute genetic data, therefore, greatly affect downstream analyses. This study aims to (1) compare the genetic variance in a single-nucleotide polymorphism panel of soybean with missing data imputed using various methods, (2) evaluate the imputation accuracy and post-imputation quality associated with these methods, and (3) evaluate the impact of imputation method on heritability and the accuracy of genome-wide prediction of soybean traits. The imputation methods we evaluated were as follows: multivariate mixed model, hidden Markov model, logical algorithm, k-nearest neighbor, single value decomposition, and random forest. We used raw genotypes from the SoyNAM project and the following phenotypes: plant height, days to maturity, grain yield, and seed protein composition.
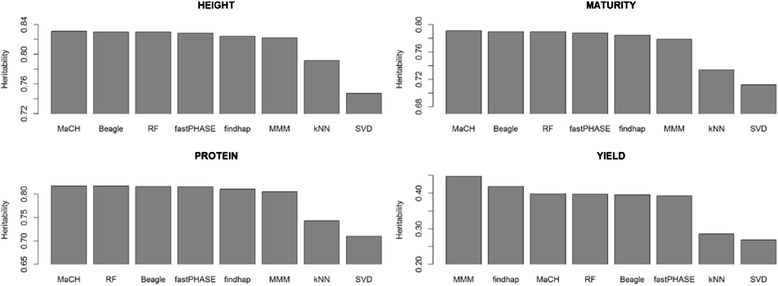
We propose an imputation method based on multivariate mixed models using pedigree information. Our methods comparison indicate that heritability of traits can be affected by the imputation method. Genotypes with missing values imputed with methods that make use of genealogic information can favor genetic analysis of highly polygenic traits, but not genome-wide prediction accuracy. The genotypic matrix captured the highest amount of genetic variance when missing loci were imputed by the method proposed in this paper.
We concluded that hidden Markov models and random forest imputation are more suitable to studies that aim analyses of highly heritable traits while pedigree-based methods can be used to best analyze traits with low heritability. Despite the notable contribution to heritability, advantages in genomic prediction were not observed by changing the imputation method. We identified significant differences across imputation methods in a dataset missing 20 % of the genotypic values. It means that genotypic data from genotyping technologies that provide a high proportion of missing values, such as GBS, should be handled carefully because the imputation method will impact downstream analysis.
全基因组关联研究和标记辅助选择的成功取决于良好的表型和基因型数据。这些数据越完整,分析结果就越有力。然而,尽管存在大量缺失数据,仍有一些下一代技术试图提供基因型信息。因此,这些技术用于推算遗传数据的程序会极大地影响下游分析。本研究旨在:(1)比较使用各种方法推算缺失数据的大豆单核苷酸多态性面板中的遗传方差;(2)评估与这些方法相关的推算准确性和推算后质量;(3)评估推算方法对大豆性状遗传力和全基因组预测准确性的影响。我们评估的推算方法如下:多变量混合模型、隐马尔可夫模型、逻辑算法、k近邻法、奇异值分解和随机森林。我们使用了来自大豆关联作图群体(SoyNAM)项目的原始基因型以及以下表型:株高、成熟天数、籽粒产量和种子蛋白组成。
我们提出了一种基于使用系谱信息的多变量混合模型的推算方法。我们的方法比较表明,性状的遗传力会受到推算方法的影响。使用系谱信息的方法推算出的缺失值基因型有利于高度多基因性状的遗传分析,但不利于全基因组预测准确性。当用本文提出的方法推算缺失位点时,基因型矩阵捕获了最高量的遗传方差。
我们得出结论,隐马尔可夫模型和随机森林推算更适合旨在分析高遗传力性状的研究,而基于系谱的方法可用于最佳分析低遗传力性状。尽管对遗传力有显著贡献,但改变推算方法未观察到在基因组预测方面的优势。我们在一个缺失20%基因型值的数据集中发现了不同推算方法之间的显著差异。这意味着来自提供高比例缺失值的基因分型技术(如GBS)的基因型数据应谨慎处理,因为推算方法会影响下游分析。