Pettengill Emily A, Pettengill James B, Binet Rachel
Division of Microbiology, Office of Regulatory Science, U.S. Food and Drug Administration, Center for Food Safety and Applied Nutrition College Park, MD, USA.
Division of Public Health Informatics and Analytics, Office of Analytics and Outreach, U.S. Food and Drug Administration, Center for Food Safety and Applied Nutrition College Park, MD, USA.
Front Microbiol. 2016 Jan 19;6:1573. doi: 10.3389/fmicb.2015.01573. eCollection 2015.
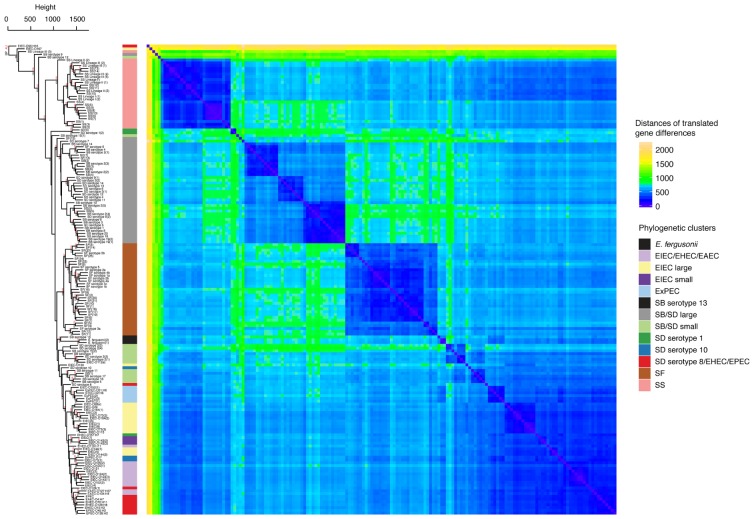
As a leading cause of bacterial dysentery, Shigella represents a significant threat to public health and food safety. Related, but often overlooked, enteroinvasive Escherichia coli (EIEC) can also cause dysentery. Current typing methods have limited ability to identify and differentiate between these pathogens despite the need for rapid and accurate identification of pathogens for clinical treatment and outbreak response. We present a comprehensive phylogeny of Shigella and EIEC using whole genome sequencing of 169 samples, constituting unparalleled strain diversity, and observe a lack of monophyly between Shigella and EIEC and among Shigella taxonomic groups. The evolutionary relationships in the phylogeny are supported by analyses of population structure and hierarchical clustering patterns of translated gene homolog abundance. Lastly, we identified a panel of 404 single nucleotide polymorphism (SNP) markers specific to each phylogenetic cluster for more accurate identification of Shigella and EIEC. Our findings show that Shigella and EIEC are not distinct evolutionary groups within the E. coli genus and, thus, EIEC as a group is not the ancestor to Shigella. The multiple analyses presented provide evidence for reconsidering the taxonomic placement of Shigella. The SNP markers offer more discriminatory power to molecular epidemiological typing methods involving these bacterial pathogens.
作为细菌性痢疾的主要病因,志贺氏菌对公众健康和食品安全构成重大威胁。相关但常被忽视的侵袭性大肠杆菌(EIEC)也可引起痢疾。尽管临床治疗和疫情应对需要快速准确地鉴定病原体,但目前的分型方法在识别和区分这些病原体方面能力有限。我们通过对169个样本进行全基因组测序,呈现了志贺氏菌和EIEC的全面系统发育,涵盖了无与伦比的菌株多样性,并观察到志贺氏菌和EIEC之间以及志贺氏菌分类群之间缺乏单系性。系统发育中的进化关系得到了群体结构分析和翻译基因同源物丰度的层次聚类模式分析的支持。最后,我们为每个系统发育簇鉴定了一组404个单核苷酸多态性(SNP)标记,以更准确地鉴定志贺氏菌和EIEC。我们的研究结果表明,志贺氏菌和EIEC在大肠杆菌属内并非不同的进化群体,因此,EIEC作为一个群体不是志贺氏菌的祖先。所呈现的多项分析为重新考虑志贺氏菌的分类地位提供了证据。SNP标记为涉及这些细菌病原体的分子流行病学分型方法提供了更强的鉴别力。