Nsiah-Sefaa Abena, McKenzie Matthew
Centre for Genetic Diseases, Hudson Institute of Medical Research, Clayton, Victoria 3168, Australia The Department of Molecular and Translational Science, Monash University, Clayton, Victoria 3168, Australia.
Centre for Genetic Diseases, Hudson Institute of Medical Research, Clayton, Victoria 3168, Australia The Department of Molecular and Translational Science, Monash University, Clayton, Victoria 3168, Australia
Biosci Rep. 2016 Feb 2;36(2):e00313. doi: 10.1042/BSR20150295.
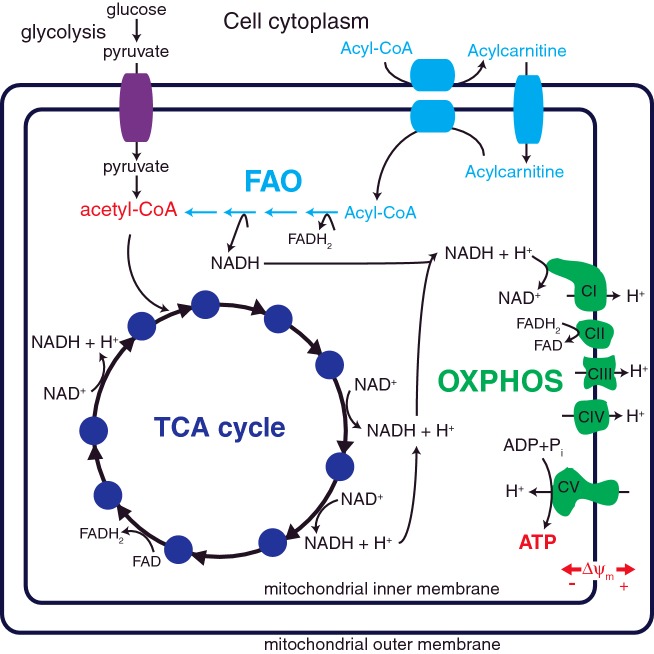
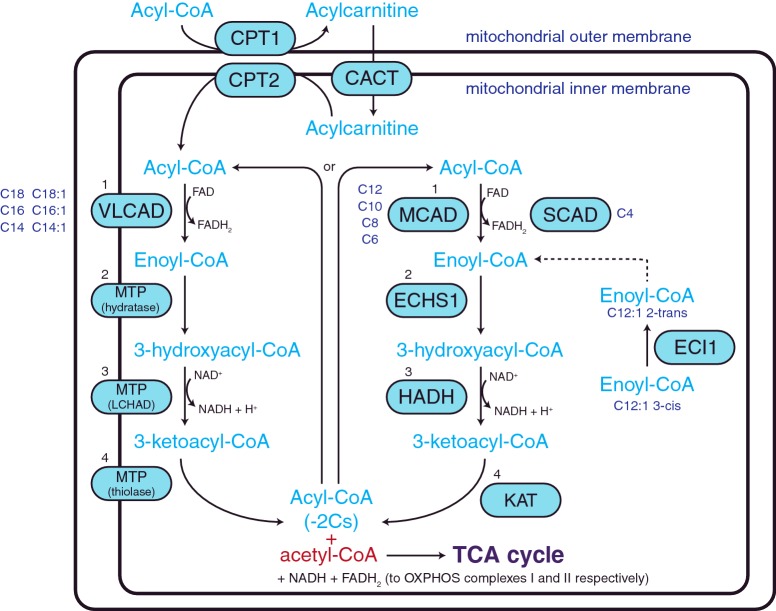
Mitochondria provide the main source of energy to eukaryotic cells, oxidizing fats and sugars to generate ATP. Mitochondrial fatty acid β-oxidation (FAO) and oxidative phosphorylation (OXPHOS) are two metabolic pathways which are central to this process. Defects in these pathways can result in diseases of the brain, skeletal muscle, heart and liver, affecting approximately 1 in 5000 live births. There are no effective therapies for these disorders, with quality of life severely reduced for most patients. The pathology underlying many aspects of these diseases is not well understood; for example, it is not clear why some patients with primary FAO deficiencies exhibit secondary OXPHOS defects. However, recent findings suggest that physical interactions exist between FAO and OXPHOS proteins, and that these interactions are critical for both FAO and OXPHOS function. Here, we review our current understanding of the interactions between FAO and OXPHOS proteins and how defects in these two metabolic pathways contribute to mitochondrial disease pathogenesis.
线粒体为真核细胞提供主要能量来源,通过氧化脂肪和糖类来生成三磷酸腺苷(ATP)。线粒体脂肪酸β-氧化(FAO)和氧化磷酸化(OXPHOS)是这一过程的两个核心代谢途径。这些途径的缺陷可导致脑、骨骼肌、心脏和肝脏疾病,约每5000例活产中就有1例受影响。目前尚无针对这些疾病的有效治疗方法,大多数患者的生活质量严重下降。这些疾病许多方面的病理机制尚未完全明确;例如,尚不清楚为何一些原发性FAO缺陷患者会出现继发性OXPHOS缺陷。然而,最近的研究结果表明,FAO和OXPHOS蛋白之间存在物理相互作用,且这些相互作用对FAO和OXPHOS功能均至关重要。在此,我们综述了目前对FAO和OXPHOS蛋白之间相互作用的理解,以及这两个代谢途径中的缺陷如何导致线粒体疾病的发病机制。