Centre for Genetic Diseases, Hudson Institute of Medical Research, 3168, Melbourne, Australia.
Department of Molecular and Translational Science, Monash University, 3168, Melbourne, Australia.
Sci Rep. 2018 Jan 9;8(1):153. doi: 10.1038/s41598-017-18530-4.
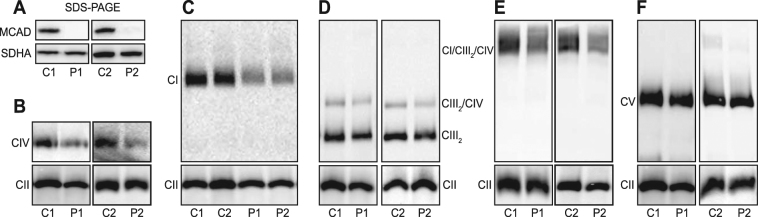
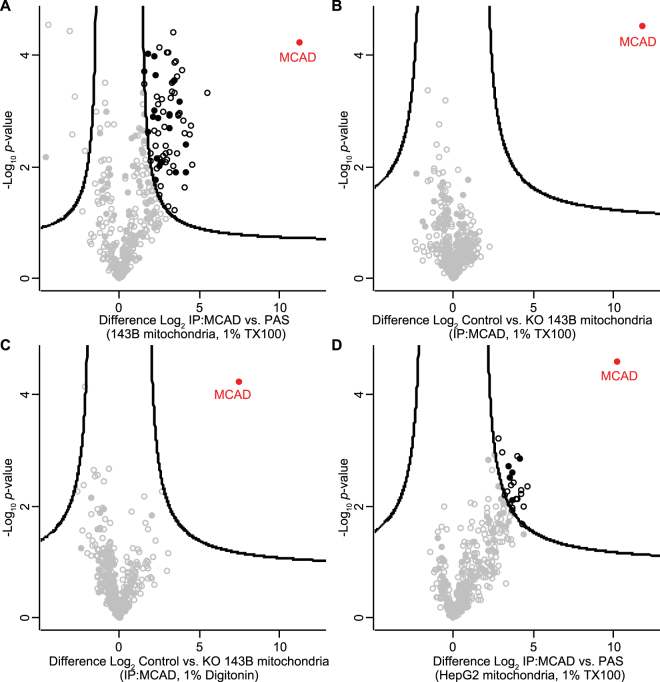
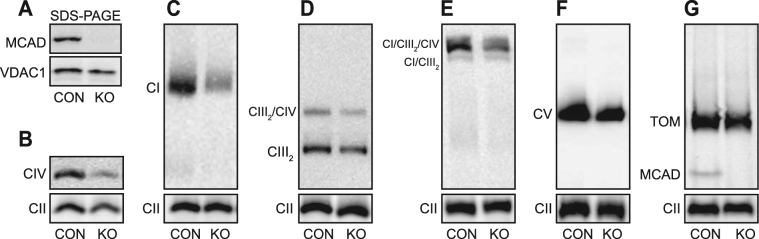
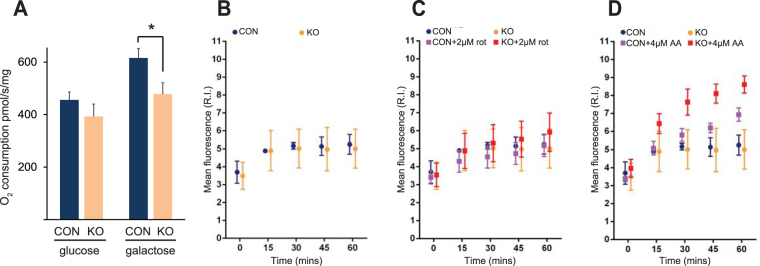
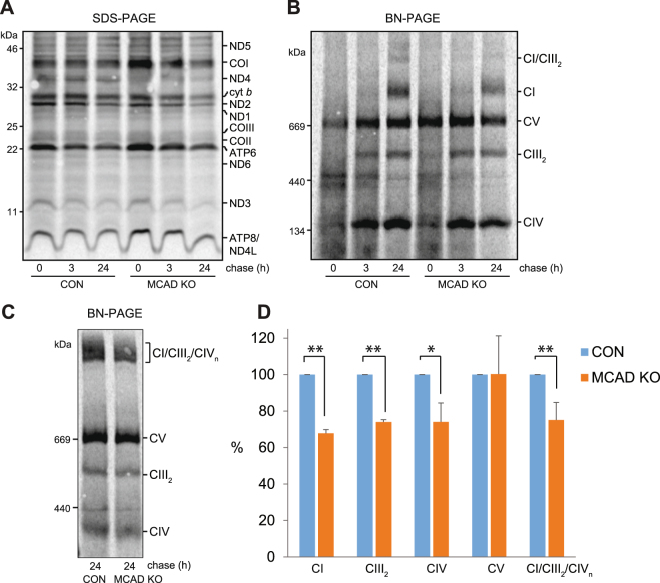
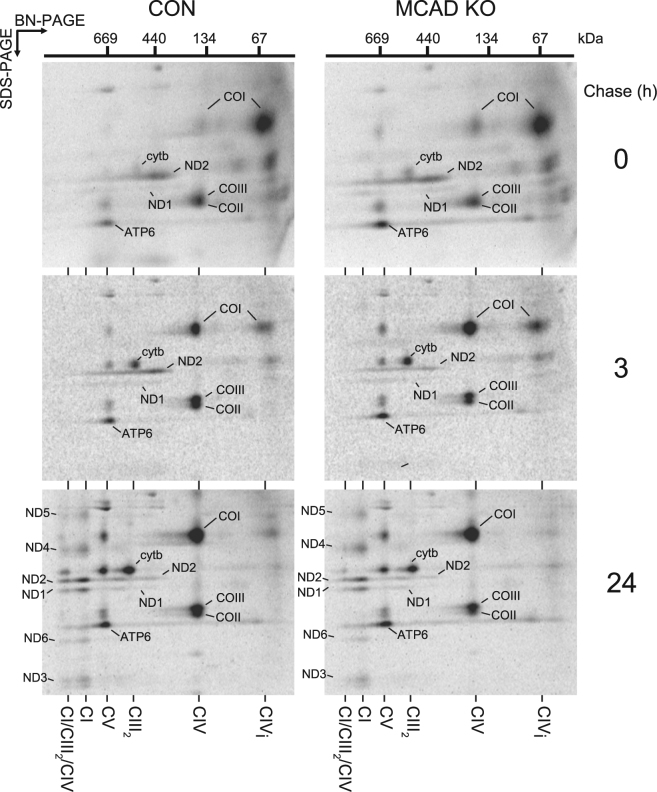
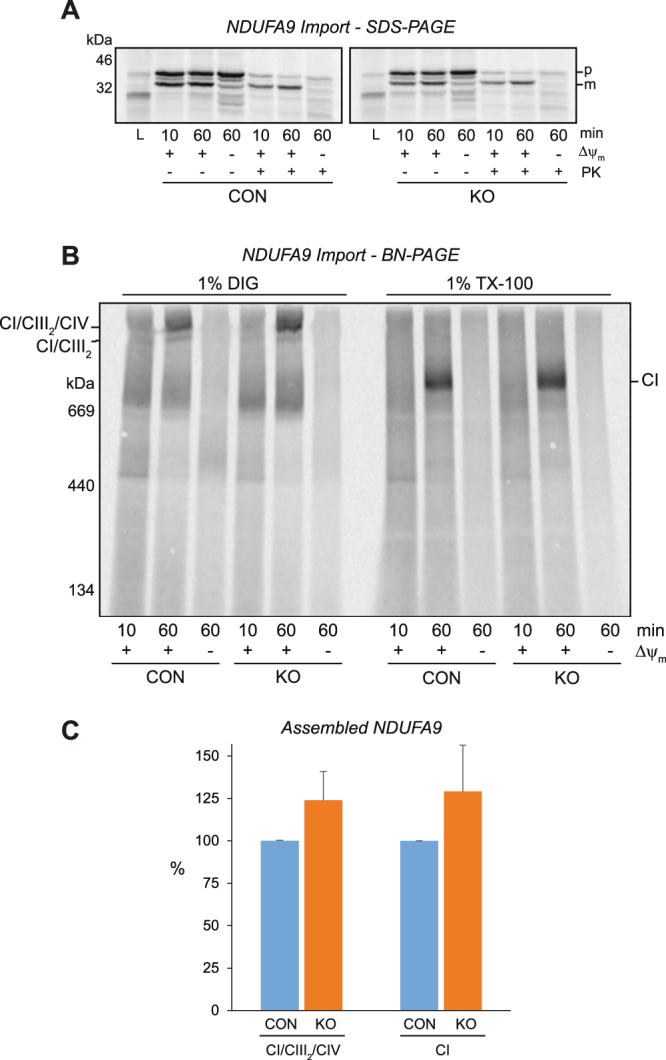
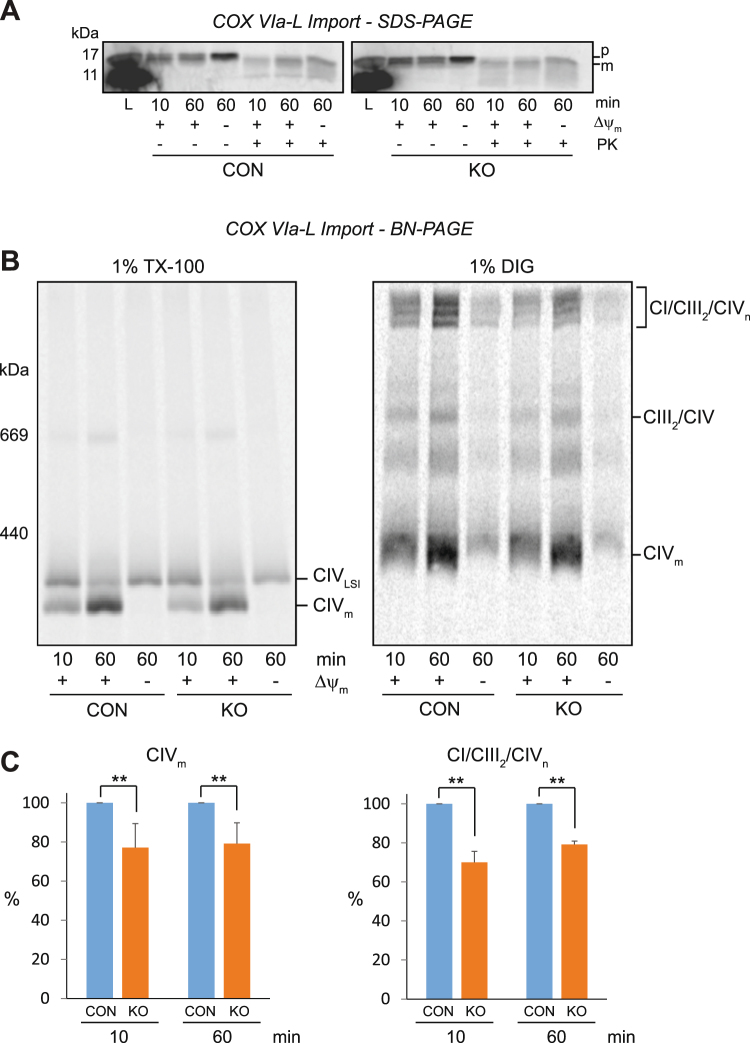
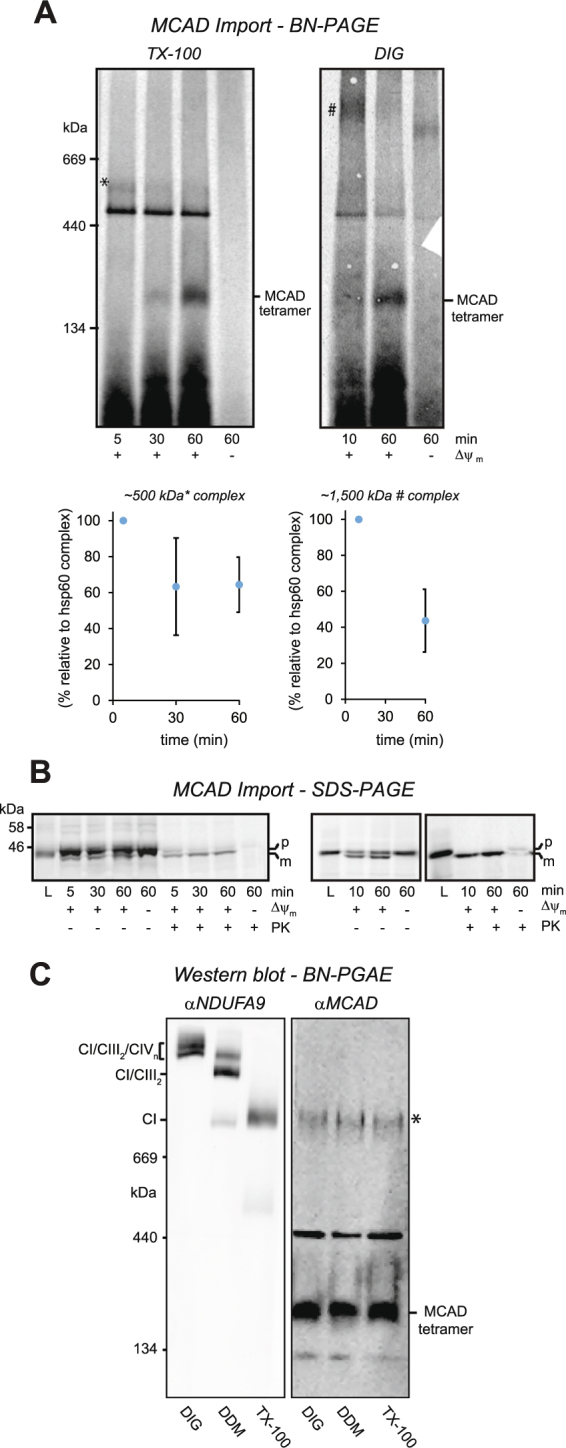
Medium-chain acyl-Coenzyme A dehydrogenase (MCAD) is involved in the initial step of mitochondrial fatty acid β-oxidation (FAO). Loss of function results in MCAD deficiency, a disorder that usually presents in childhood with hypoketotic hypoglycemia, vomiting and lethargy. While the disruption of mitochondrial fatty acid metabolism is the primary metabolic defect, secondary defects in mitochondrial oxidative phosphorylation (OXPHOS) may also contribute to disease pathogenesis. Therefore, we examined OXPHOS activity and stability in MCAD-deficient patient fibroblasts that have no detectable MCAD protein. We found a deficit in mitochondrial oxygen consumption, with reduced steady-state levels of OXPHOS complexes I, III and IV, as well as the OXPHOS supercomplex. To examine the mechanisms involved, we generated an MCAD knockout (KO) using human 143B osteosarcoma cells. These cells also exhibited defects in OXPHOS complex function and steady-state levels, as well as disrupted biogenesis of newly-translated OXPHOS subunits. Overall, our findings suggest that the loss of MCAD is associated with a reduction in steady-state OXPHOS complex levels, resulting in secondary defects in OXPHOS function which may contribute to the pathology of MCAD deficiency.
中链酰基辅酶 A 脱氢酶(MCAD)参与线粒体脂肪酸β氧化(FAO)的初始步骤。功能丧失导致 MCAD 缺乏症,这是一种通常在儿童时期出现的疾病,表现为低酮性低血糖、呕吐和嗜睡。虽然线粒体脂肪酸代谢的破坏是主要的代谢缺陷,但线粒体氧化磷酸化(OXPHOS)的继发性缺陷也可能导致疾病发病机制。因此,我们检查了没有检测到 MCAD 蛋白的 MCAD 缺陷患者成纤维细胞中的 OXPHOS 活性和稳定性。我们发现线粒体耗氧量不足,OXPHOS 复合物 I、III 和 IV 以及 OXPHOS 超复合物的稳态水平降低。为了研究涉及的机制,我们使用人骨肉瘤 143B 细胞生成了 MCAD 敲除(KO)。这些细胞的 OXPHOS 复合物功能和稳态水平也存在缺陷,新翻译的 OXPHOS 亚基的生物发生也受到干扰。总体而言,我们的研究结果表明,MCAD 的缺失与 OXPHOS 复合物稳态水平降低有关,导致 OXPHOS 功能的继发性缺陷,这可能导致 MCAD 缺乏症的病理。