Peng Qi, Wu Bin, Jiang Mali, Jin Jing, Hou Zhipeng, Zheng Jennifer, Zhang Jiangyang, Duan Wenzhen
Division of Neurobiology, Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore, Maryland, United States of America.
Department of General Practice, The First hospital of China Medical University, Shenyang, Liaoning Province, China.
PLoS One. 2016 Feb 9;11(2):e0148839. doi: 10.1371/journal.pone.0148839. eCollection 2016.
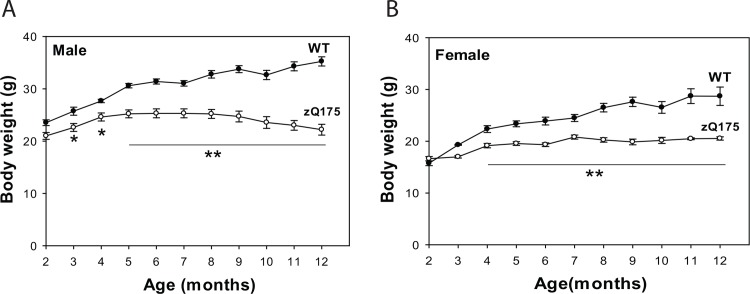
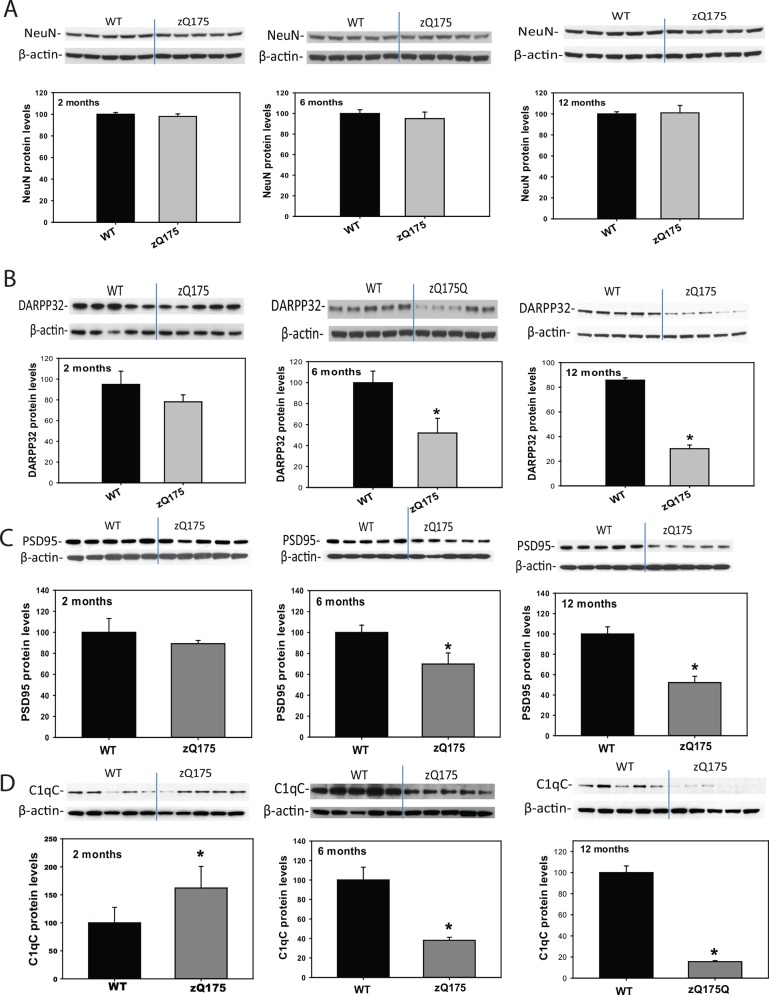
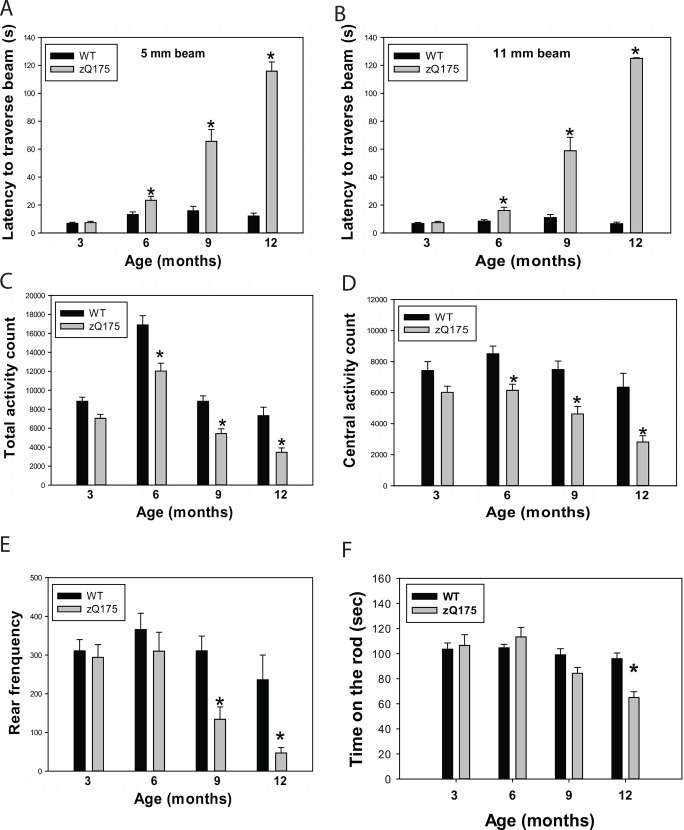
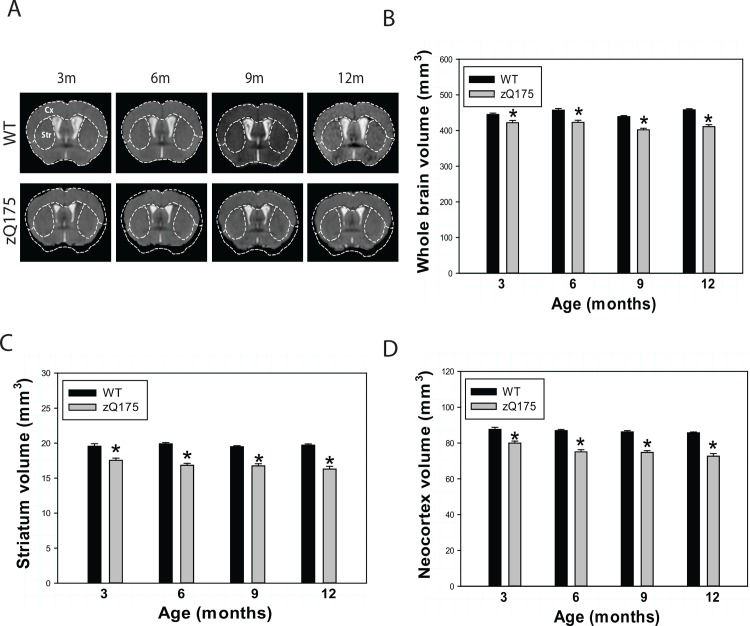
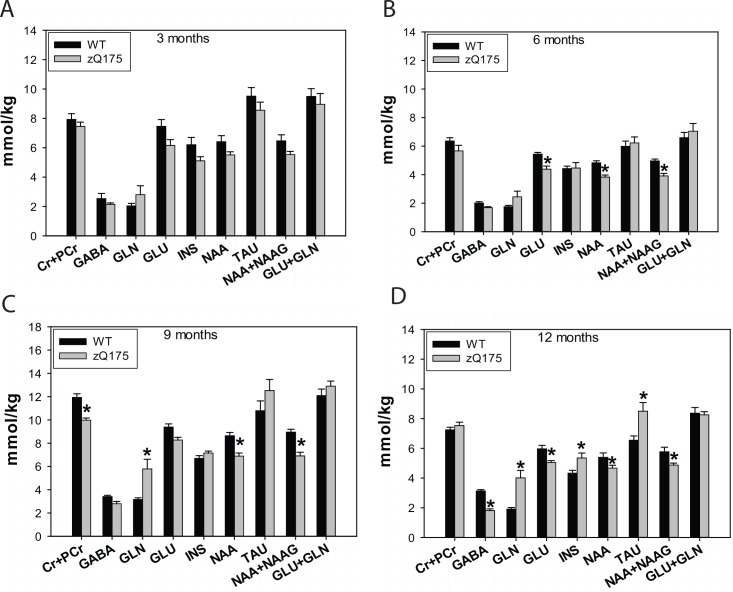
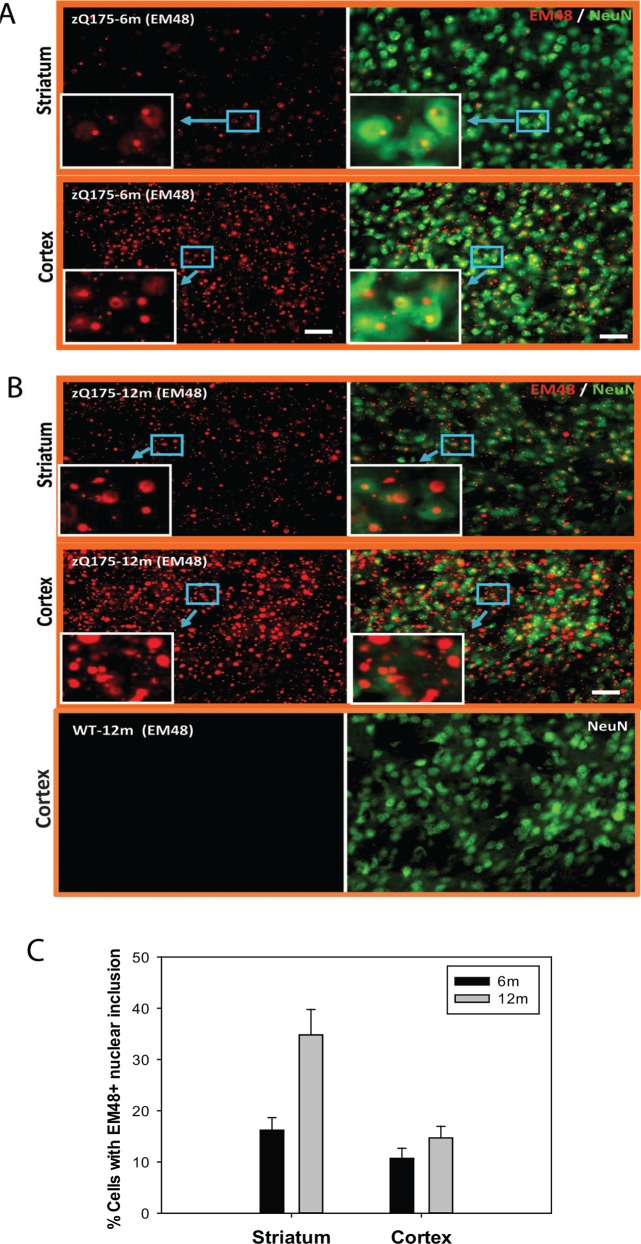
Huntington's disease (HD) is caused by an expansion of the trinucleotide poly (CAG) tract located in exon 1 of the huntingtin (Htt) gene leading to progressive neurodegeneration in selected brain regions, and associated functional impairments in motor, cognitive, and psychiatric domains. Since the discovery of the gene mutation that causes the disease, mouse models have been developed by different strategies. Recently, a new model, the zQ175 knock-in (KI) line, was developed in an attempt to have the Htt gene in a context and causing a phenotype that more closely mimics HD in humans. The behavioral phenotype was characterized across the independent laboratories and important features reminiscent of human HD are observed in zQ175 mice. In the current study, we characterized the zQ175 model housed in an academic laboratory under reversed dark-light cycle, including motor function, in vivo longitudinal structural MRI imaging for brain volume, MRS for striatal metabolites, neuropathology, as well as a panel of key disease marker proteins in the striatum at different ages. Our results suggest that homozygous zQ175 mice exhibited significant brain atrophy before the motor deficits and brain metabolite changes. Altered striatal medium spiny neuronal marker, postsynaptic marker protein and complement component C1qC also characterized zQ175 mice. Our results confirmed that the zQ175 KI model is valuable in understanding of HD-like pathophysiology and evaluation of potential therapeutics. Our data also provide suggestions to select appropriate outcome measurements in preclinical studies using the zQ175 mice.
亨廷顿舞蹈症(HD)是由位于亨廷顿蛋白(Htt)基因外显子1中的三核苷酸聚(CAG)序列扩增引起的,导致特定脑区的进行性神经变性,以及运动、认知和精神领域的相关功能损害。自从发现导致该疾病的基因突变以来,已经通过不同策略开发了小鼠模型。最近,开发了一种新的模型,即zQ175基因敲入(KI)品系,试图使Htt基因处于一种环境中,并产生一种更接近人类HD的表型。在各个独立实验室对其行为表型进行了表征,并且在zQ175小鼠中观察到了让人联想到人类HD的重要特征。在本研究中,我们对饲养在学术实验室中、处于颠倒明暗周期下的zQ175模型进行了表征,包括运动功能、用于脑容量的体内纵向结构MRI成像、用于纹状体代谢物的磁共振波谱分析、神经病理学,以及不同年龄纹状体中一组关键疾病标志物蛋白。我们的结果表明,纯合zQ175小鼠在出现运动缺陷和脑代谢物变化之前就表现出明显的脑萎缩。纹状体中等棘状神经元标志物、突触后标志物蛋白和补体成分C1qC的改变也是zQ175小鼠的特征。我们的结果证实,zQ175 KI模型在理解HD样病理生理学和评估潜在治疗方法方面具有重要价值。我们的数据还为在使用zQ175小鼠的临床前研究中选择合适的结果测量指标提供了建议。