Dellicour Simon, Rose Rebecca, Pybus Oliver G
Department of Zoology, University of Oxford, Oxford, OX1 3PS, UK.
Rega Institute for Medical Research, Clinical and Epidemiological Virology, Department of Microbiology and Immunology, KU Leuven, University of Leuven, Minderbroedersstaat 10, 3000, Leuven, Belgium.
BMC Bioinformatics. 2016 Feb 11;17:82. doi: 10.1186/s12859-016-0924-x.
Phylogenetic analysis is now an important tool in the study of viral outbreaks. It can reconstruct epidemic history when surveillance epidemiology data are sparse, and can indicate transmission linkages among infections that may not otherwise be evident. However, a remaining challenge is to develop an analytical framework that can test hypotheses about the effect of environmental variables on pathogen spatial spread. Recent phylogeographic approaches can reconstruct the history of virus dispersal from sampled viral genomes and infer the locations of ancestral infections. Such methods provide a unique source of spatio-temporal information, and are exploited here.
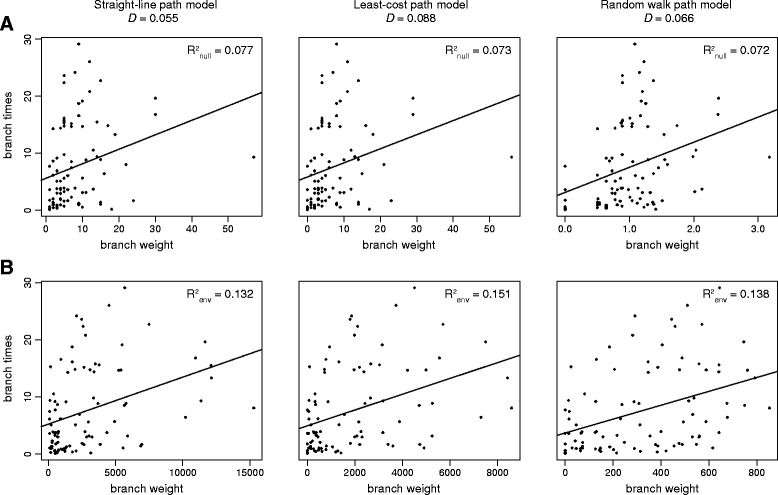
We present and apply a new statistical framework that combines genomic and geographic data to test the impact of environmental variables on the mode and tempo of pathogen dispersal during emerging epidemics. First, the spatial history of an emerging pathogen is estimated using standard phylogeographic methods. The inferred dispersal path for each phylogenetic lineage is then assigned a "weight" using environmental data (e.g. altitude, land cover). Next, tests measure the association between each environmental variable and lineage movement. A randomisation procedure is used to assess statistical confidence and we validate this approach using simulated data. We apply our new framework to a set of gene sequences from an epidemic of rabies virus in North American raccoons. We test the impact of six different environmental variables on this epidemic and demonstrate that elevation is associated with a slower rabies spread in a natural population.
This study shows that it is possible to integrate genomic and environmental data in order to test hypotheses concerning the mode and tempo of virus dispersal during emerging epidemics.
系统发育分析如今是病毒爆发研究中的一项重要工具。当监测流行病学数据匮乏时,它能够重构疫情历史,并且能够揭示那些在其他情况下可能不明显的感染之间的传播联系。然而,一个尚存的挑战是开发一个分析框架,该框架能够检验关于环境变量对病原体空间传播影响的假设。近期的系统发育地理学方法能够从采样的病毒基因组重构病毒传播历史,并推断祖先感染的位置。此类方法提供了独特的时空信息来源,本文对此加以利用。
我们提出并应用了一个新的统计框架,该框架结合基因组和地理数据来检验环境变量对新发疫情期间病原体传播模式和速度的影响。首先,使用标准的系统发育地理学方法估计新发病原体的空间历史。然后,利用环境数据(如海拔、土地覆盖)为每个系统发育谱系推断出的传播路径赋予一个“权重”。接下来,通过检验测量每个环境变量与谱系移动之间的关联。使用随机化程序评估统计置信度,并利用模拟数据验证该方法。我们将新框架应用于一组北美浣熊狂犬病病毒疫情的基因序列。我们检验了六种不同环境变量对此次疫情的影响,并证明海拔与狂犬病在自然种群中的传播速度较慢有关。
本研究表明,整合基因组和环境数据以检验关于新发疫情期间病毒传播模式和速度的假设是可行的。