Gregor Ivan, Dröge Johannes, Schirmer Melanie, Quince Christopher, McHardy Alice C
Max-Planck Research Group for Computational Genomics and Epidemiology, Max-Planck Institute for Informatics, Saarbrücken, Germany; Department of Algorithmic Bioinformatics, Heinrich-Heine-University Düsseldorf, Düsseldorf, Germany; Computational Biology of Infection Research, Helmholtz Center for Infection Research, Braunschweig, Germany.
The Broad Institute of MIT and Harvard , Cambridge, MA , United States.
PeerJ. 2016 Feb 8;4:e1603. doi: 10.7717/peerj.1603. eCollection 2016.
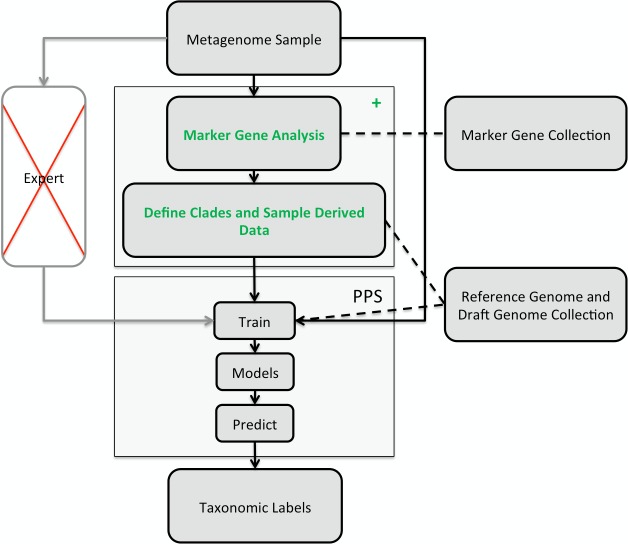
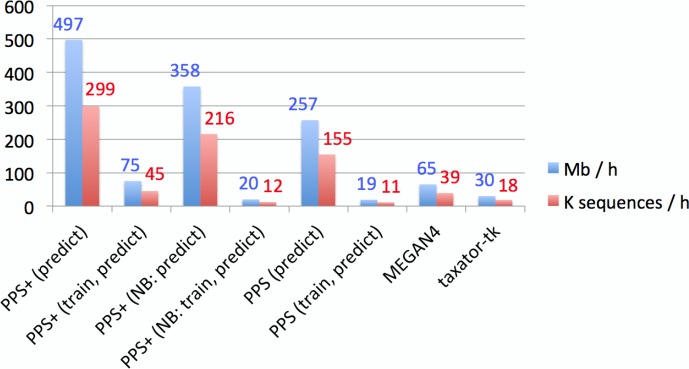
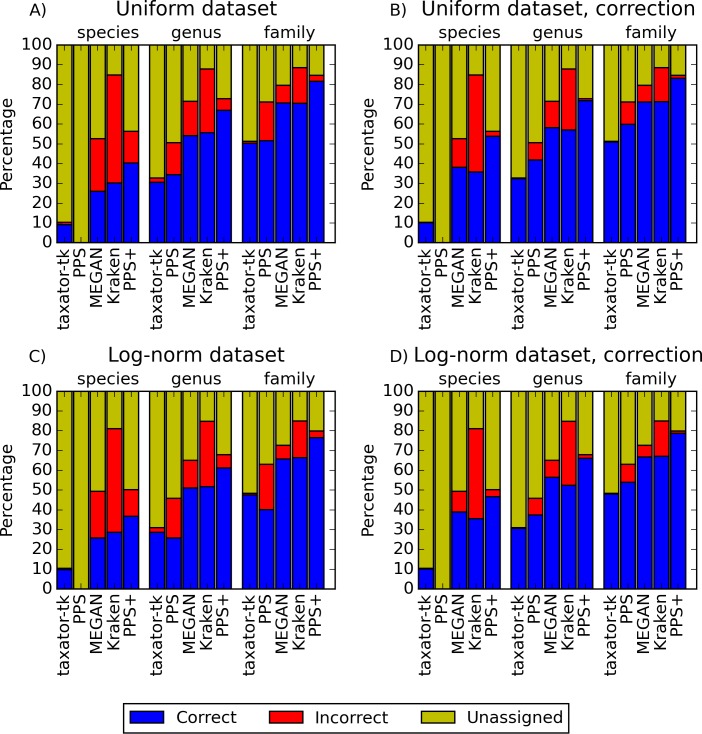
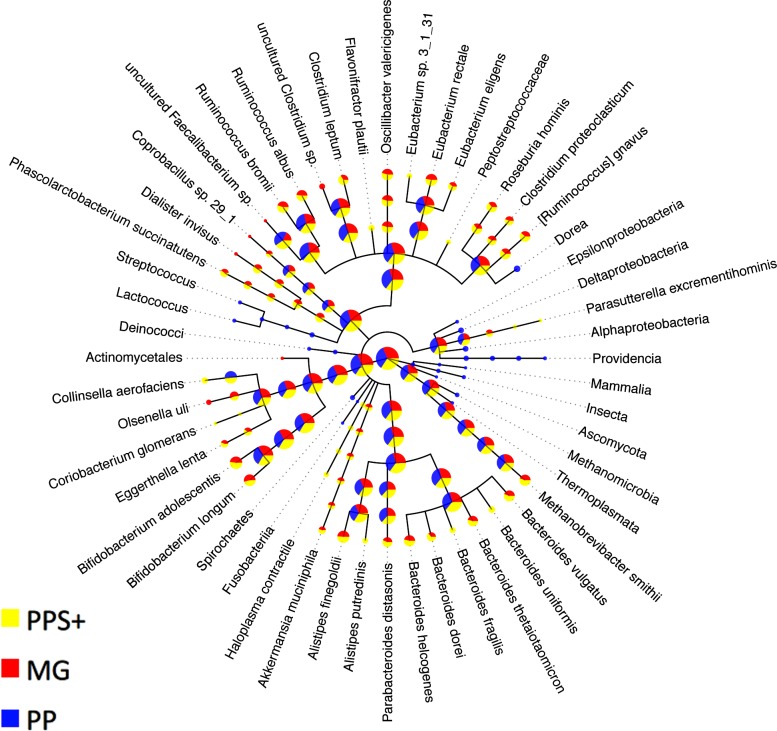
Background. Metagenomics is an approach for characterizing environmental microbial communities in situ, it allows their functional and taxonomic characterization and to recover sequences from uncultured taxa. This is often achieved by a combination of sequence assembly and binning, where sequences are grouped into 'bins' representing taxa of the underlying microbial community. Assignment to low-ranking taxonomic bins is an important challenge for binning methods as is scalability to Gb-sized datasets generated with deep sequencing techniques. One of the best available methods for species bins recovery from deep-branching phyla is the expert-trained PhyloPythiaS package, where a human expert decides on the taxa to incorporate in the model and identifies 'training' sequences based on marker genes directly from the sample. Due to the manual effort involved, this approach does not scale to multiple metagenome samples and requires substantial expertise, which researchers who are new to the area do not have. Results. We have developed PhyloPythiaS+, a successor to our PhyloPythia(S) software. The new (+) component performs the work previously done by the human expert. PhyloPythiaS+ also includes a new k-mer counting algorithm, which accelerated the simultaneous counting of 4-6-mers used for taxonomic binning 100-fold and reduced the overall execution time of the software by a factor of three. Our software allows to analyze Gb-sized metagenomes with inexpensive hardware, and to recover species or genera-level bins with low error rates in a fully automated fashion. PhyloPythiaS+ was compared to MEGAN, taxator-tk, Kraken and the generic PhyloPythiaS model. The results showed that PhyloPythiaS+ performs especially well for samples originating from novel environments in comparison to the other methods. Availability. PhyloPythiaS+ in a virtual machine is available for installation under Windows, Unix systems or OS X on: https://github.com/algbioi/ppsp/wiki.
背景。宏基因组学是一种原位表征环境微生物群落的方法,它能够对微生物群落进行功能和分类特征分析,并从未培养的分类群中恢复序列。这通常通过序列组装和分箱相结合来实现,即将序列分组到代表潜在微生物群落分类群的“箱”中。对于分箱方法而言,将序列分配到低等级分类箱以及扩展到由深度测序技术生成的千兆字节大小数据集都是一项重大挑战。从深分支门类中恢复物种箱的最佳可用方法之一是经过专家训练的PhyloPythiaS软件包,其中由人类专家决定纳入模型的分类群,并基于直接来自样本的标记基因识别“训练”序列。由于涉及人工操作,这种方法无法扩展到多个宏基因组样本,并且需要大量专业知识,而该领域的新手研究人员并不具备这些知识。结果。我们开发了PhyloPythiaS+,它是我们的PhyloPythia(S)软件的后续版本。新的(+)组件执行了之前由人类专家完成的工作。PhyloPythiaS+还包括一种新的k-mer计数算法,该算法将用于分类分箱的4至6-mer的同时计数速度提高了100倍,并将软件的总体执行时间缩短了三分之一。我们的软件允许使用低成本硬件分析千兆字节大小的宏基因组,并以完全自动化的方式以低错误率恢复物种或属水平的箱。将PhyloPythiaS+与MEGAN、taxator-tk、Kraken和通用的PhyloPythiaS模型进行了比较。结果表明,与其他方法相比,PhyloPythiaS+对于源自新环境的样本表现尤其出色。可用性。可在以下网址获取虚拟机中的PhyloPythiaS+,以便在Windows、Unix系统或OS X上安装:https://github.com/algbioi/ppsp/wiki。