Boutet Gilles, Alves Carvalho Susete, Falque Matthieu, Peterlongo Pierre, Lhuillier Emeline, Bouchez Olivier, Lavaud Clément, Pilet-Nayel Marie-Laure, Rivière Nathalie, Baranger Alain
INRA, UMR 1349 IGEPP, BP35327, Le Rheu Cedex, 35653, France.
PISOM, UMT INRA/CETIOM, BP35327, Le Rheu Cedex, 35653, France.
BMC Genomics. 2016 Feb 18;17:121. doi: 10.1186/s12864-016-2447-2.
Progress in genetics and breeding in pea still suffers from the limited availability of molecular resources. SNP markers that can be identified through affordable sequencing processes, without the need for prior genome reduction or a reference genome to assemble sequencing data would allow the discovery and genetic mapping of thousands of molecular markers. Such an approach could significantly speed up genetic studies and marker assisted breeding for non-model species.
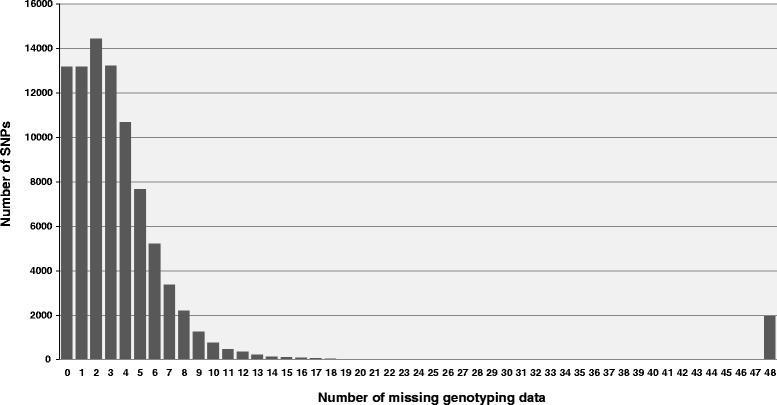
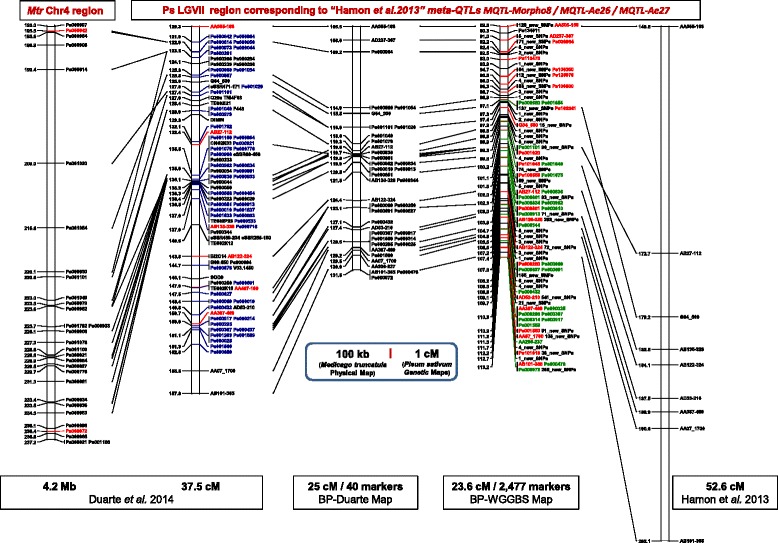
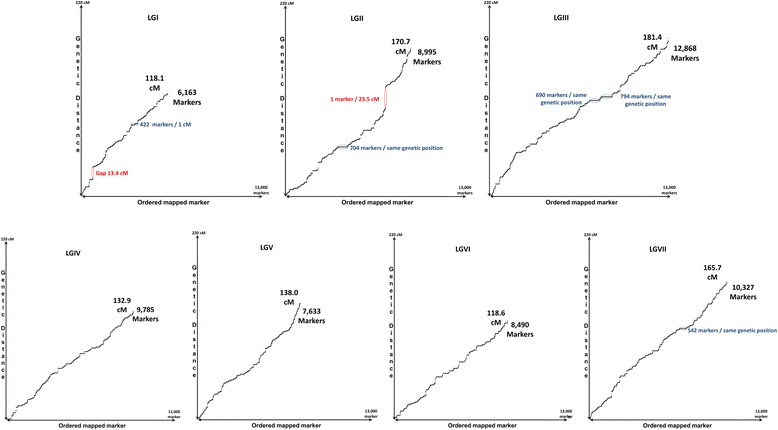
A total of 419,024 SNPs were discovered using HiSeq whole genome sequencing of four pea lines, followed by direct identification of SNP markers without assembly using the discoSnp tool. Subsequent filtering led to the identification of 131,850 highly designable SNPs, polymorphic between at least two of the four pea lines. A subset of 64,754 SNPs was called and genotyped by short read sequencing on a subpopulation of 48 RILs from the cross 'Baccara' x 'PI180693'. This data was used to construct a WGGBS-derived pea genetic map comprising 64,263 markers. This map is collinear with previous pea consensus maps and therefore with the Medicago truncatula genome. Sequencing of four additional pea lines showed that 33 % to 64 % of the mapped SNPs, depending on the pairs of lines considered, are polymorphic and can therefore be useful in other crosses. The subsequent genotyping of a subset of 1000 SNPs, chosen for their mapping positions using a KASP™ assay, showed that almost all generated SNPs are highly designable and that most (95 %) deliver highly qualitative genotyping results. Using rather low sequencing coverages in SNP discovery and in SNP inferring did not hinder the identification of hundreds of thousands of high quality SNPs.
The development and optimization of appropriate tools in SNP discovery and genetic mapping have allowed us to make available a massive new genomic resource in pea. It will be useful for both fine mapping within chosen QTL confidence intervals and marker assisted breeding for important traits in pea improvement.
豌豆的遗传学和育种进展仍受分子资源可用性有限的制约。通过经济实惠的测序流程即可识别的单核苷酸多态性(SNP)标记,无需事先进行基因组简化或参考基因组来组装测序数据,这将有助于发现数千个分子标记并进行遗传定位。这种方法可显著加快非模式物种的遗传研究和标记辅助育种进程。
利用HiSeq对四个豌豆品系进行全基因组测序,共发现419,024个SNP,随后使用discoSnp工具直接识别SNP标记而无需组装。后续筛选鉴定出131,850个高度可设计的SNP,在四个豌豆品系中的至少两个之间具有多态性。通过对杂交组合‘Baccara’בPI180693’的48个重组自交系(RIL)亚群体进行短读长测序,对64,754个SNP子集进行了分型和基因分型。该数据用于构建包含64,263个标记的基于全基因组亚硫酸氢盐测序(WGGBS)的豌豆遗传图谱。该图谱与先前的豌豆共识图谱共线,因此也与蒺藜苜蓿基因组共线。对另外四个豌豆品系的测序表明,根据所考虑的品系对,33%至64%的已定位SNP具有多态性,因此可用于其他杂交组合。随后使用竞争性等位基因特异性PCR(KASP™)分析对1000个SNP子集进行基因分型,这些SNP是根据其定位位置选择的,结果表明几乎所有生成的SNP都是高度可设计的,并且大多数(95%)提供高度定性的基因分型结果。在SNP发现和SNP推断中使用相对较低的测序覆盖度并不妨碍鉴定出数十万高质量的SNP。
在SNP发现和遗传图谱构建中适当工具的开发和优化,使我们能够在豌豆中提供大量新的基因组资源。这对于在选定的数量性状基因座(QTL)置信区间内进行精细定位以及在豌豆改良中对重要性状进行标记辅助育种都将是有用的。