Gori Kevin, Suchan Tomasz, Alvarez Nadir, Goldman Nick, Dessimoz Christophe
European Molecular Biology Laboratory, European Bioinformatics Institute, Wellcome Trust Campus, Hinxton, United Kingdom.
Department of Ecology and Evolution, Biophore Building, UNIL-Sorge, University of Lausanne, Lausanne, Switzerland.
Mol Biol Evol. 2016 Jun;33(6):1590-605. doi: 10.1093/molbev/msw038. Epub 2016 Feb 17.
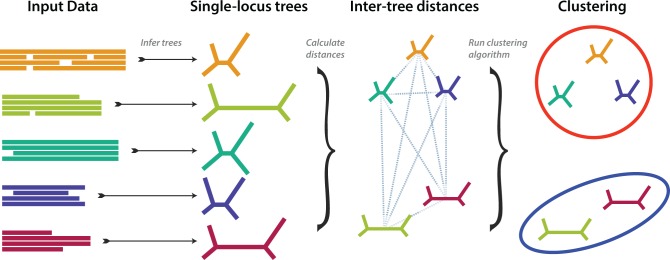
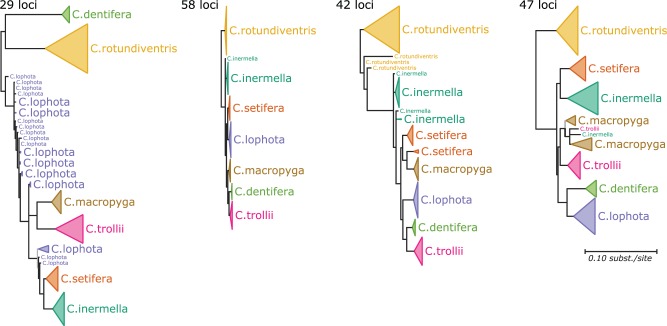
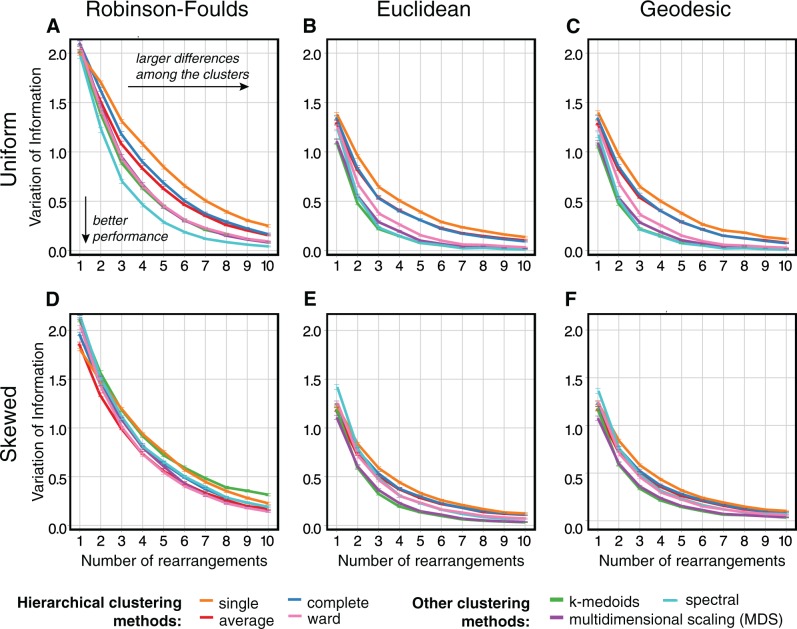
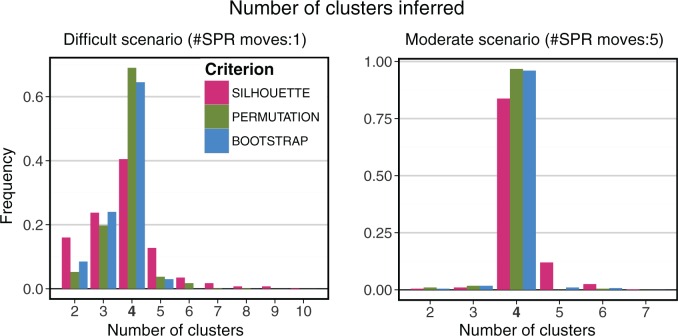
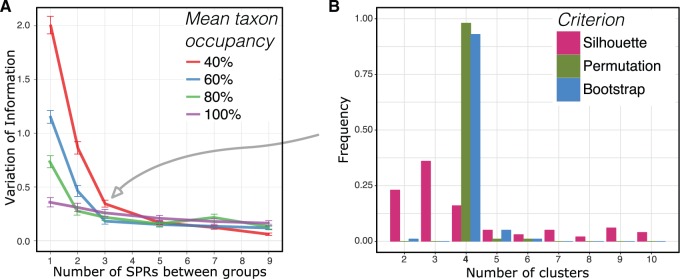
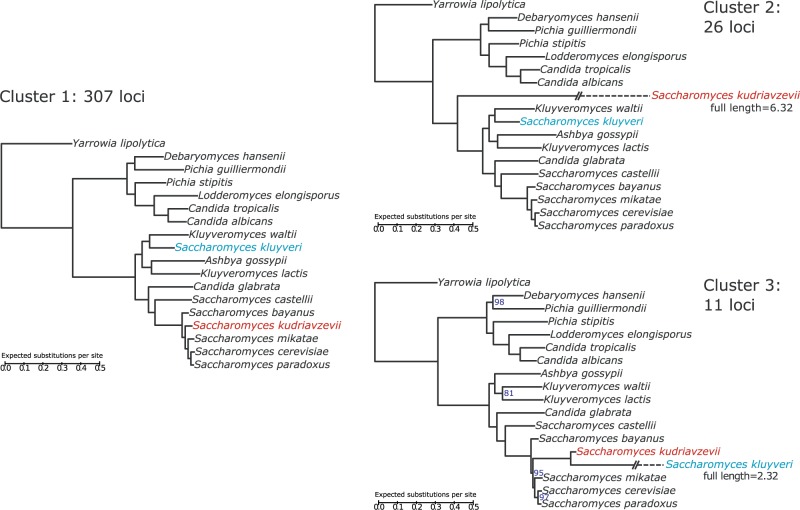
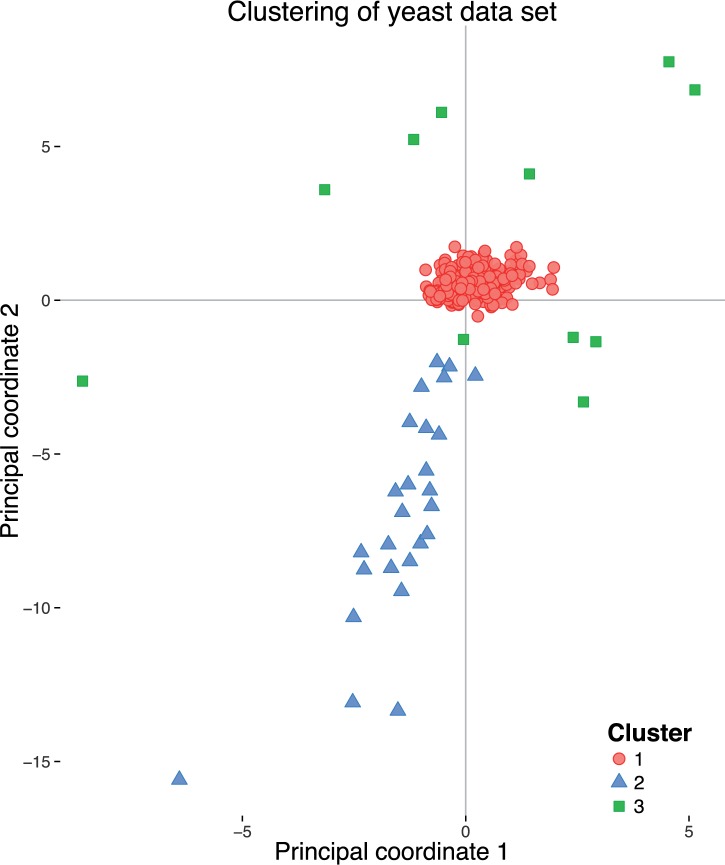
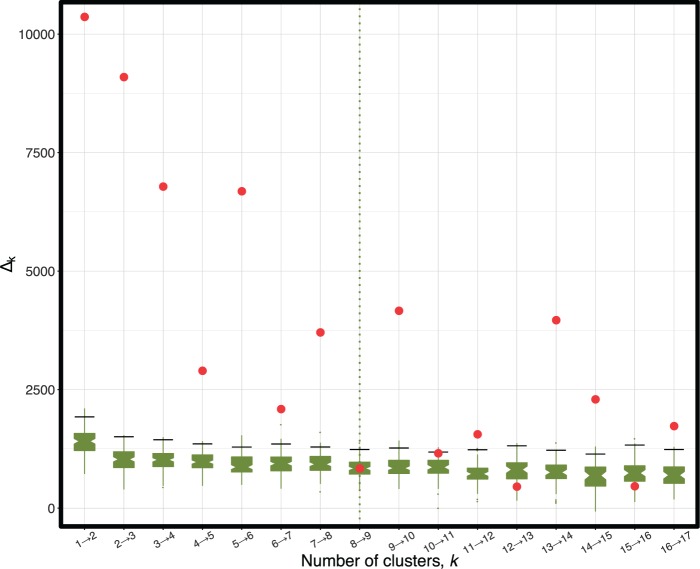
Phylogenetic inference can potentially result in a more accurate tree using data from multiple loci. However, if the loci are incongruent-due to events such as incomplete lineage sorting or horizontal gene transfer-it can be misleading to infer a single tree. To address this, many previous contributions have taken a mechanistic approach, by modeling specific processes. Alternatively, one can cluster loci without assuming how these incongruencies might arise. Such "process-agnostic" approaches typically infer a tree for each locus and cluster these. There are, however, many possible combinations of tree distance and clustering methods; their comparative performance in the context of tree incongruence is largely unknown. Furthermore, because standard model selection criteria such as AIC cannot be applied to problems with a variable number of topologies, the issue of inferring the optimal number of clusters is poorly understood. Here, we perform a large-scale simulation study of phylogenetic distances and clustering methods to infer loci of common evolutionary history. We observe that the best-performing combinations are distances accounting for branch lengths followed by spectral clustering or Ward's method. We also introduce two statistical tests to infer the optimal number of clusters and show that they strongly outperform the silhouette criterion, a general-purpose heuristic. We illustrate the usefulness of the approach by 1) identifying errors in a previous phylogenetic analysis of yeast species and 2) identifying topological incongruence among newly sequenced loci of the globeflower fly genus Chiastocheta We release treeCl, a new program to cluster genes of common evolutionary history (http://git.io/treeCl).
系统发育推断使用多个基因座的数据可能会得到一棵更准确的树。然而,如果基因座不一致——由于不完全谱系分选或水平基因转移等事件——推断一棵单一的树可能会产生误导。为了解决这个问题,许多先前的研究采用了机械方法,即对特定过程进行建模。或者,人们可以在不假设这些不一致如何产生的情况下对基因座进行聚类。这种“与过程无关”的方法通常会为每个基因座推断一棵树并对这些树进行聚类。然而,树距离和聚类方法有许多可能的组合;它们在树不一致情况下的比较性能在很大程度上是未知的。此外,由于标准的模型选择标准(如AIC)不能应用于拓扑数量可变的问题,因此对推断最佳聚类数的问题了解甚少。在这里,我们对系统发育距离和聚类方法进行了大规模模拟研究,以推断具有共同进化历史的基因座。我们观察到,表现最佳的组合是考虑分支长度的距离,其次是光谱聚类或沃德方法。我们还引入了两种统计检验来推断最佳聚类数,并表明它们的性能明显优于轮廓系数这一通用启发式方法。我们通过1)识别先前酵母物种系统发育分析中的错误,以及2)识别金莲花蝇属Chiastocheta新测序基因座之间的拓扑不一致,来说明该方法的有用性。我们发布了treeCl,一个用于聚类具有共同进化历史的基因的新程序(http://git.io/treeCl)。