Merlet Benjamin, Paulhe Nils, Vinson Florence, Frainay Clément, Chazalviel Maxime, Poupin Nathalie, Gloaguen Yoann, Giacomoni Franck, Jourdan Fabien
TOXALIM (Research Centre in Food Toxicology), Institut National de la Recherche Agronomique, UMR1331, Université de Toulouse Toulouse, France.
Nutrition Humaine, Plateforme d'Exploration du Métabolisme, Institut National de la Recherche Agronomique, Centre Clermont-Ferrand-Theix, UMR 1019 Saint-Genès-Champanelle, France.
Front Mol Biosci. 2016 Feb 16;3:2. doi: 10.3389/fmolb.2016.00002. eCollection 2016.
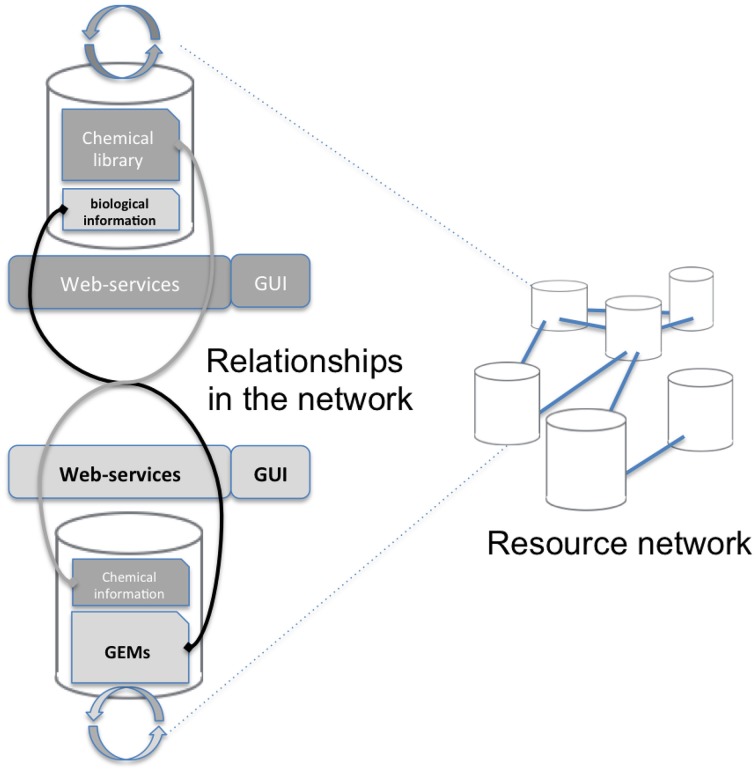
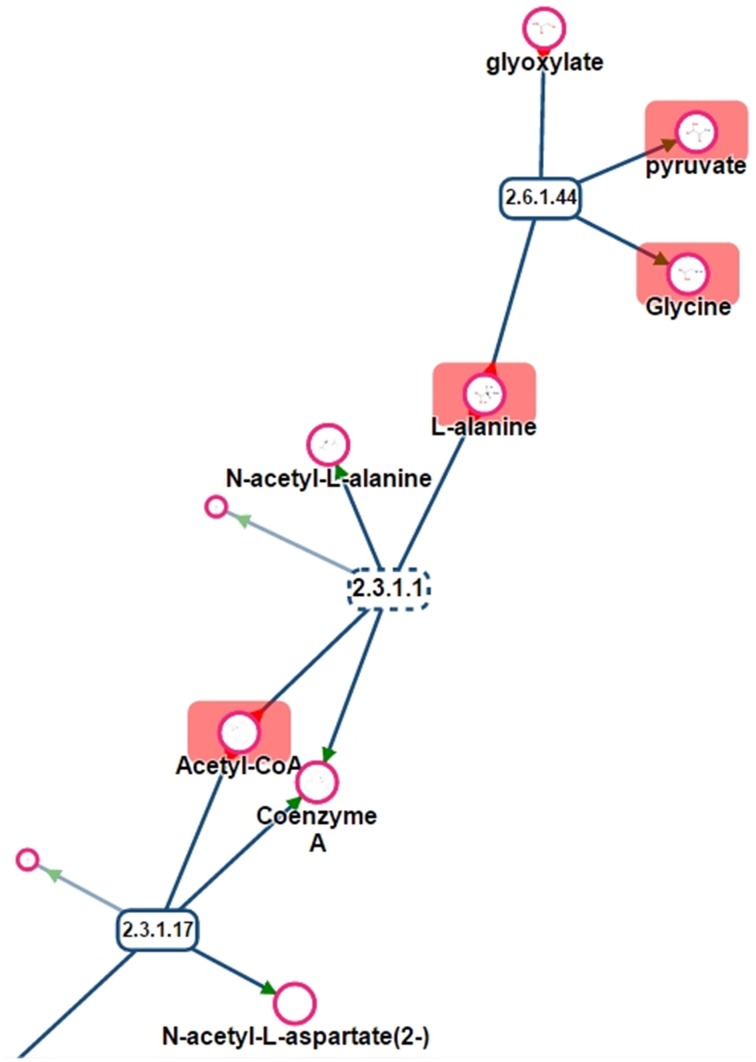
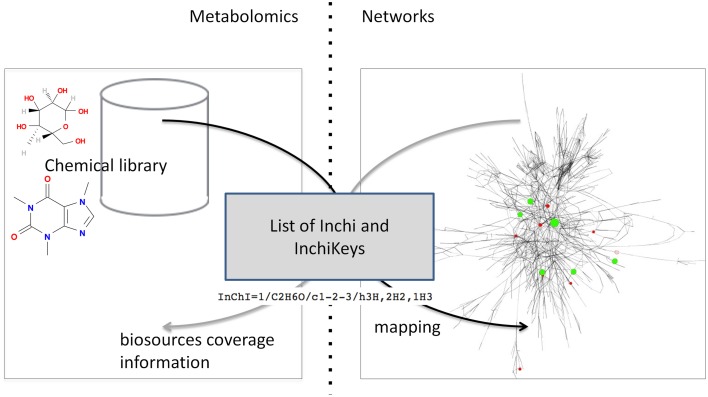
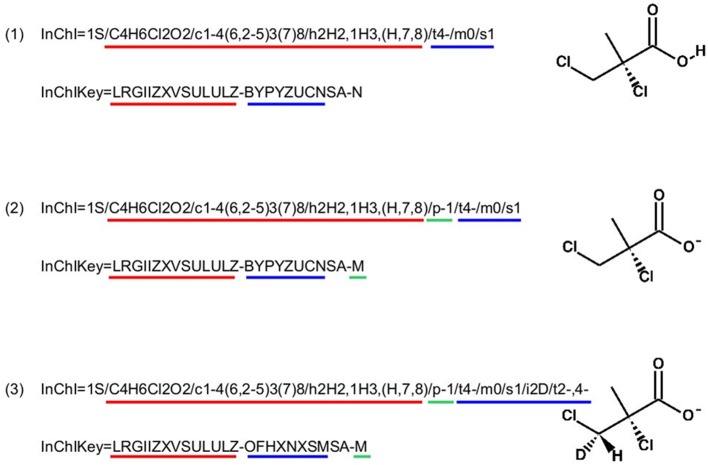
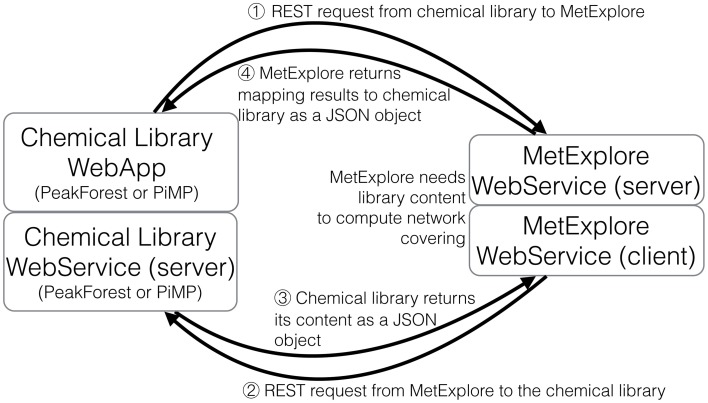
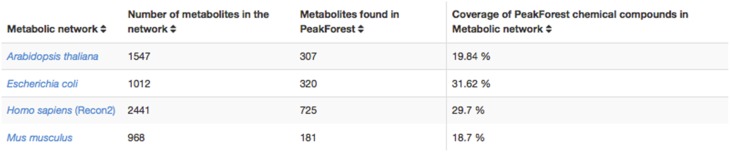
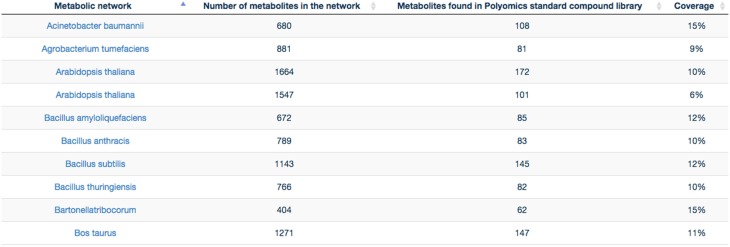
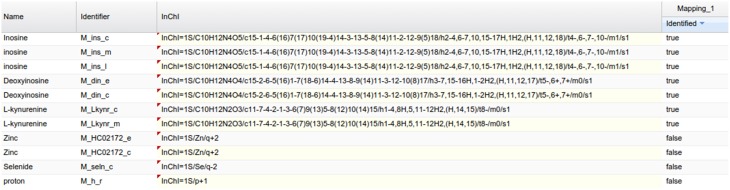
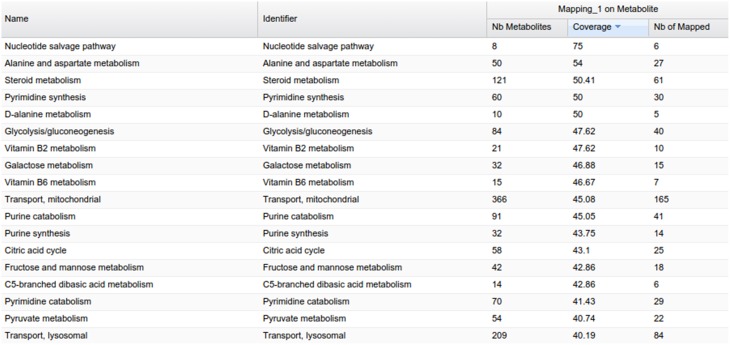
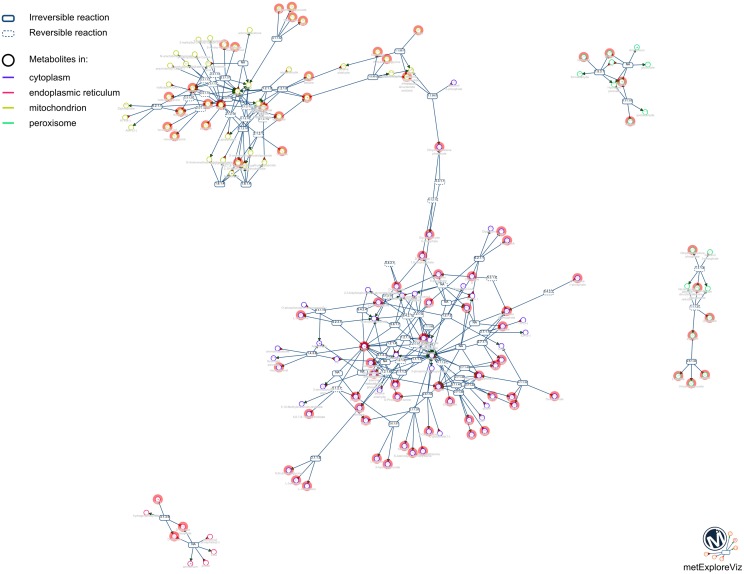
This article describes a generic programmatic method for mapping chemical compound libraries on organism-specific metabolic networks from various databases (KEGG, BioCyc) and flat file formats (SBML and Matlab files). We show how this pipeline was successfully applied to decipher the coverage of chemical libraries set up by two metabolomics facilities MetaboHub (French National infrastructure for metabolomics and fluxomics) and Glasgow Polyomics (GP) on the metabolic networks available in the MetExplore web server. The present generic protocol is designed to formalize and reduce the volume of information transfer between the library and the network database. Matching of metabolites between libraries and metabolic networks is based on InChIs or InChIKeys and therefore requires that these identifiers are specified in both libraries and networks. In addition to providing covering statistics, this pipeline also allows the visualization of mapping results in the context of metabolic networks. In order to achieve this goal, we tackled issues on programmatic interaction between two servers, improvement of metabolite annotation in metabolic networks and automatic loading of a mapping in genome scale metabolic network analysis tool MetExplore. It is important to note that this mapping can also be performed on a single or a selection of organisms of interest and is thus not limited to large facilities.
本文介绍了一种通用的编程方法,用于将化合物库映射到来自各种数据库(KEGG、BioCyc)和平板文件格式(SBML和Matlab文件)的特定生物体代谢网络上。我们展示了该流程如何成功应用于解读由两个代谢组学设施MetaboHub(法国国家代谢组学和通量组学基础设施)和格拉斯哥多组学(GP)建立的化学库对MetExplore网络服务器中可用代谢网络的覆盖情况。当前的通用协议旨在规范并减少库与网络数据库之间的信息传输量。库与代谢网络之间代谢物的匹配基于国际化学标识符(InChIs)或国际化学标识符密钥(InChIKeys),因此要求在库和网络中都指定这些标识符。除了提供覆盖统计信息外,该流程还允许在代谢网络的背景下可视化映射结果。为了实现这一目标,我们解决了两个服务器之间的编程交互问题、代谢网络中代谢物注释的改进以及在基因组规模代谢网络分析工具MetExplore中自动加载映射的问题。需要注意的是,这种映射也可以在单个或选定的感兴趣生物体上进行,因此不限于大型设施。