Wong Nicholas C, Pope Bernard J, Candiloro Ida L, Korbie Darren, Trau Matt, Wong Stephen Q, Mikeska Thomas, Zhang Xinmin, Pitman Mark, Eggers Stefanie, Doyle Stephen R, Dobrovic Alexander
Translational Genomics and Epigenomics Laboratory, Olivia Newton-John Cancer Research Institute, Heidelberg, Victoria, 3084, Australia.
Murdoch Childrens Research Institute, The Royal Children's Hospital, Parkville, Victoria, 3052, Australia.
BMC Bioinformatics. 2016 Feb 24;17:98. doi: 10.1186/s12859-016-0950-8.
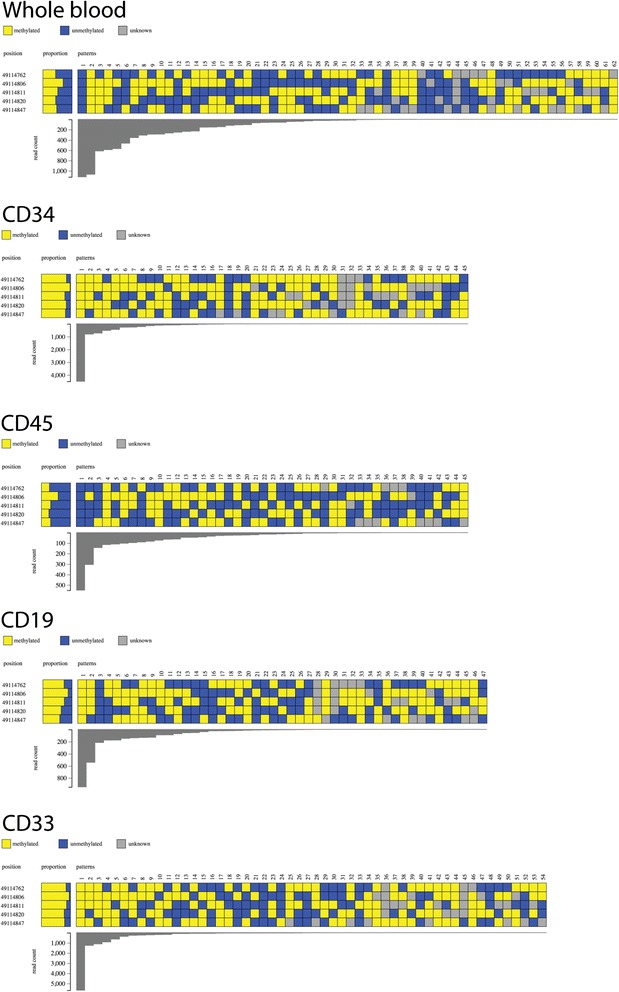
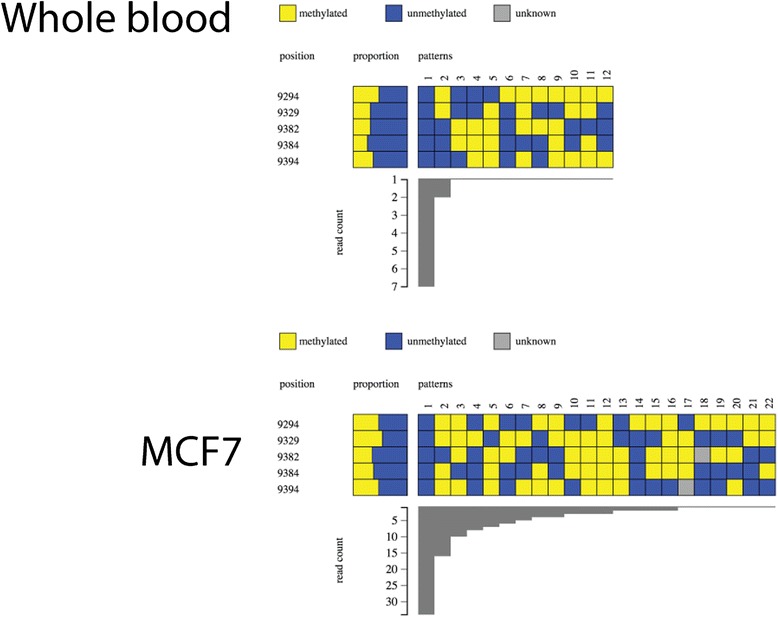
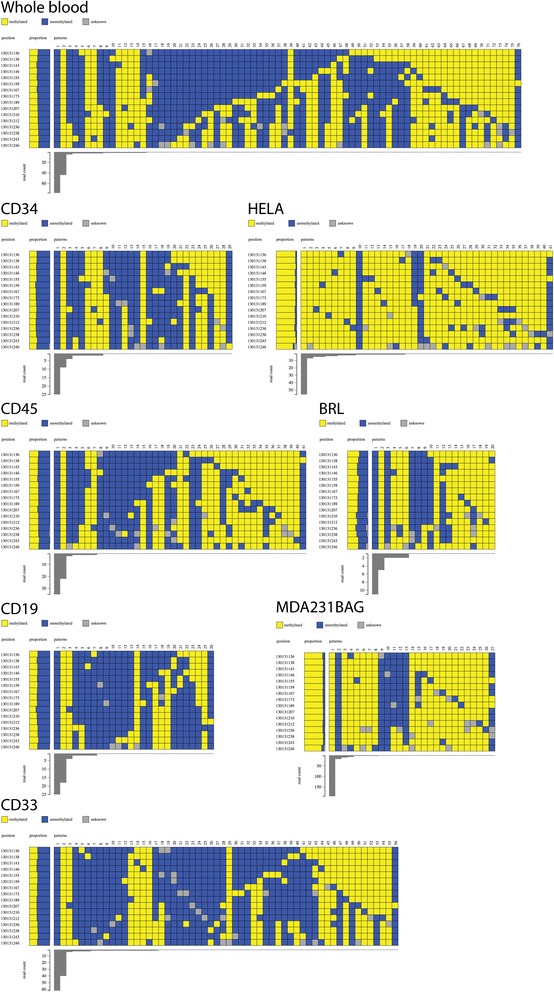
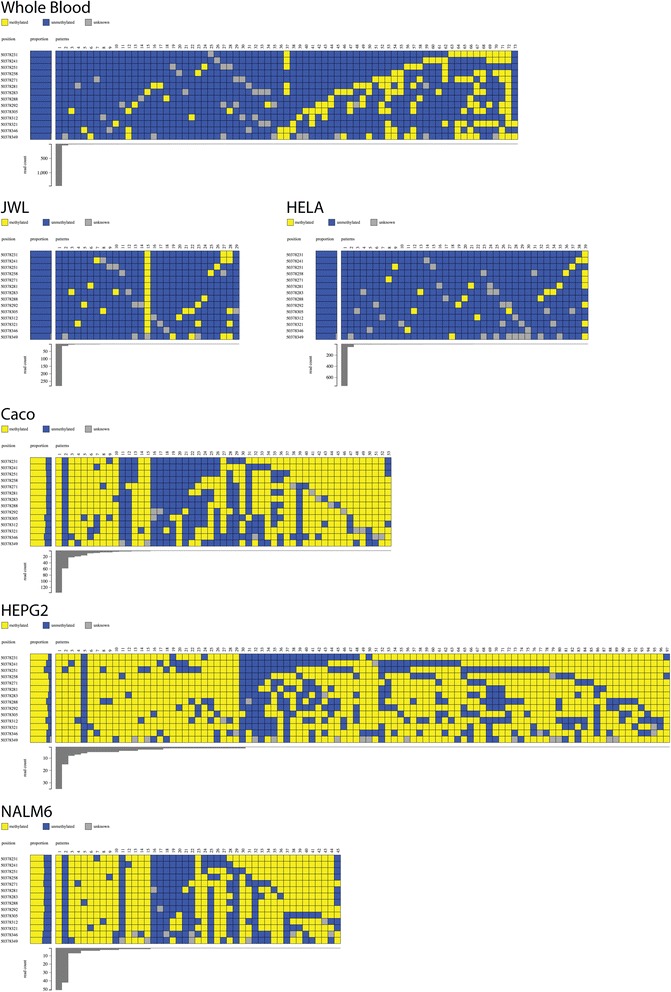
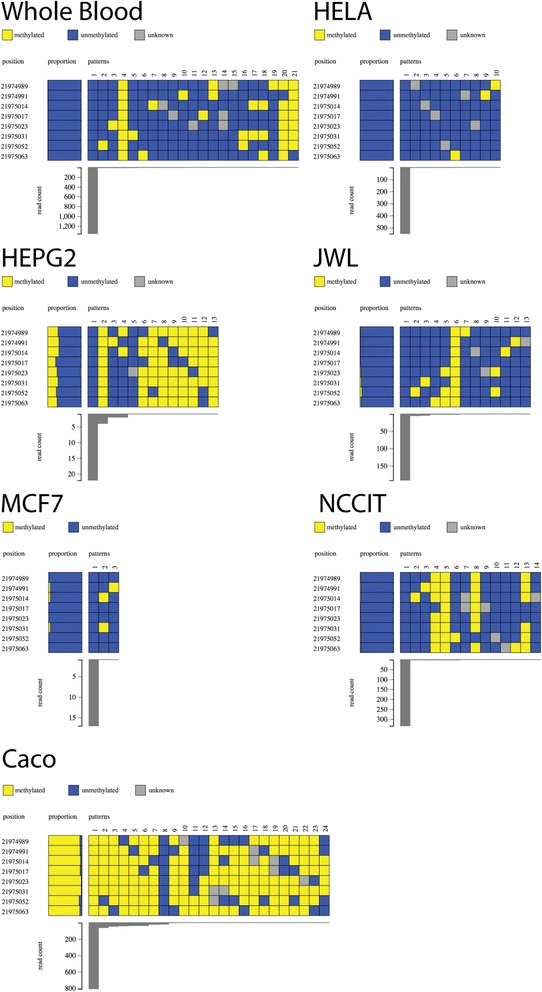
DNA methylation at a gene promoter region has the potential to regulate gene transcription. Patterns of methylation over multiple CpG sites in a region are often complex and cell type specific, with the region showing multiple allelic patterns in a sample. This complexity is commonly obscured when DNA methylation data is summarised as an average percentage value for each CpG site (or aggregated across CpG sites). True representation of methylation patterns can only be fully characterised by clonal analysis. Deep sequencing provides the ability to investigate clonal DNA methylation patterns in unprecedented detail and scale, enabling the proper characterisation of the heterogeneity of methylation patterns. However, the sheer amount and complexity of sequencing data requires new synoptic approaches to visualise the distribution of allelic patterns.
We have developed a new analysis and visualisation software tool "Methpat", that extracts and displays clonal DNA methylation patterns from massively parallel sequencing data aligned using Bismark. Methpat was used to analyse multiplex bisulfite amplicon sequencing on a range of CpG island targets across a panel of human cell lines and primary tissues. Methpat was able to represent the clonal diversity of epialleles analysed at specific gene promoter regions. We also used Methpat to describe epiallelic DNA methylation within the mitochondrial genome.
Methpat can summarise and visualise epiallelic DNA methylation results from targeted amplicon, massively parallel sequencing of bisulfite converted DNA in a compact and interpretable format. Unlike currently available tools, Methpat can visualise the diversity of epiallelic DNA methylation patterns in a sample.
基因启动子区域的DNA甲基化具有调控基因转录的潜力。一个区域内多个CpG位点的甲基化模式通常很复杂且具有细胞类型特异性,该区域在样本中呈现多种等位基因模式。当DNA甲基化数据被总结为每个CpG位点的平均百分比值(或跨CpG位点汇总)时,这种复杂性通常会被掩盖。甲基化模式的真实表征只能通过克隆分析来全面描述。深度测序能够以前所未有的细节和规模研究克隆DNA甲基化模式,从而能够正确表征甲基化模式的异质性。然而,测序数据的数量和复杂性需要新的概要方法来可视化等位基因模式的分布。
我们开发了一种新的分析和可视化软件工具“Methpat”,它从使用Bismark比对的大规模平行测序数据中提取并显示克隆DNA甲基化模式。Methpat被用于分析一系列人类细胞系和原代组织中多个CpG岛靶点的多重亚硫酸氢盐扩增子测序。Methpat能够呈现特定基因启动子区域分析的表观等位基因的克隆多样性。我们还使用Methpat来描述线粒体基因组内的表观等位基因DNA甲基化。
Methpat可以以紧凑且可解释的格式总结和可视化来自靶向扩增子、亚硫酸氢盐转化DNA的大规模平行测序的表观等位基因DNA甲基化结果。与目前可用的工具不同,Methpat可以可视化样本中表观等位基因DNA甲基化模式的多样性。