School of Computer Science and Technology, Tianjin University, Tianjin 300350, China.
Tianjin University Institute of Computational Biology, Tianjin University, Tianjin 300350, China.
Int J Mol Sci. 2018 Feb 8;19(2):511. doi: 10.3390/ijms19020511.
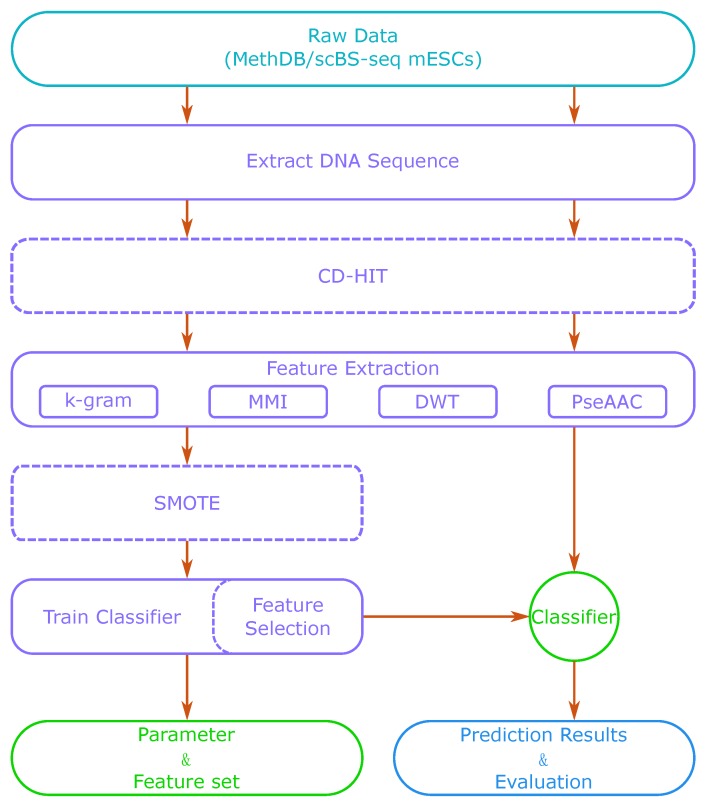
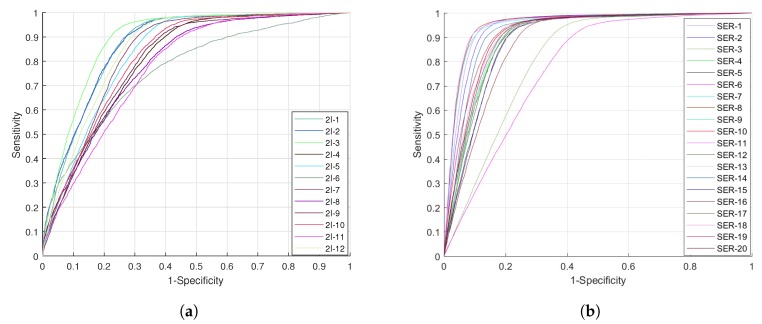
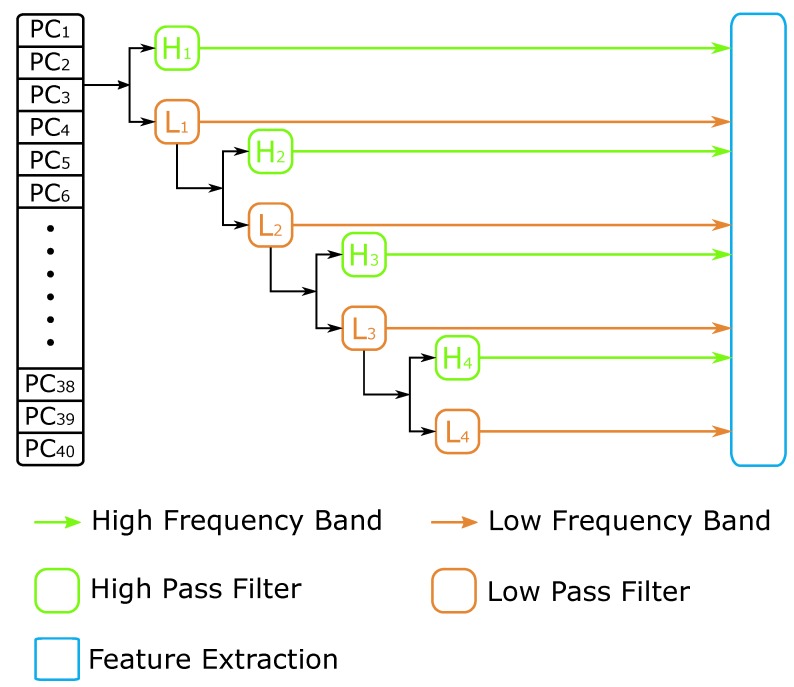
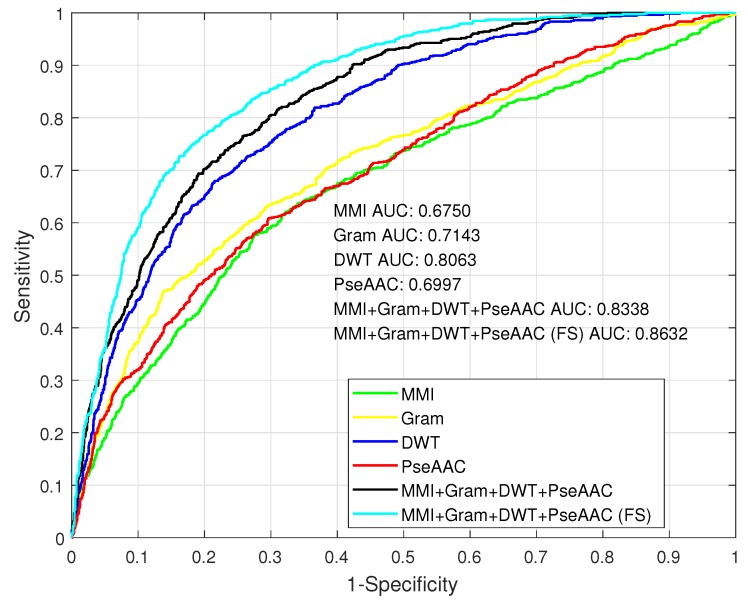
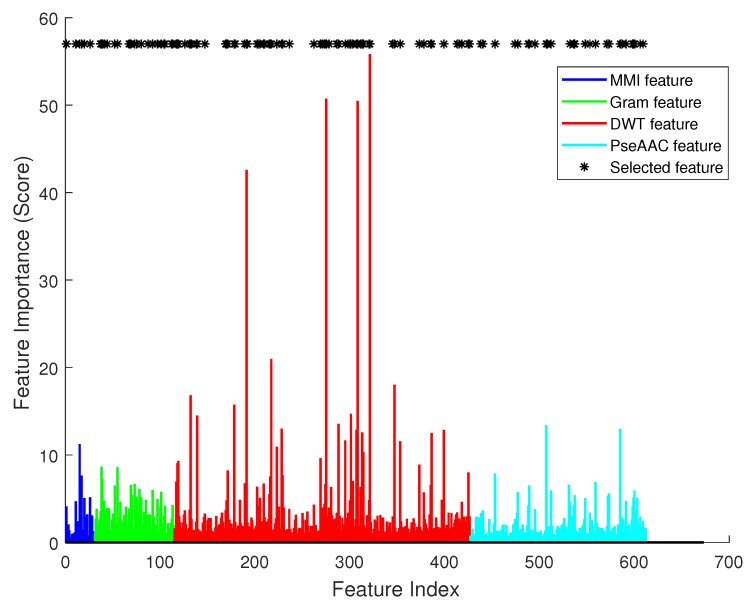
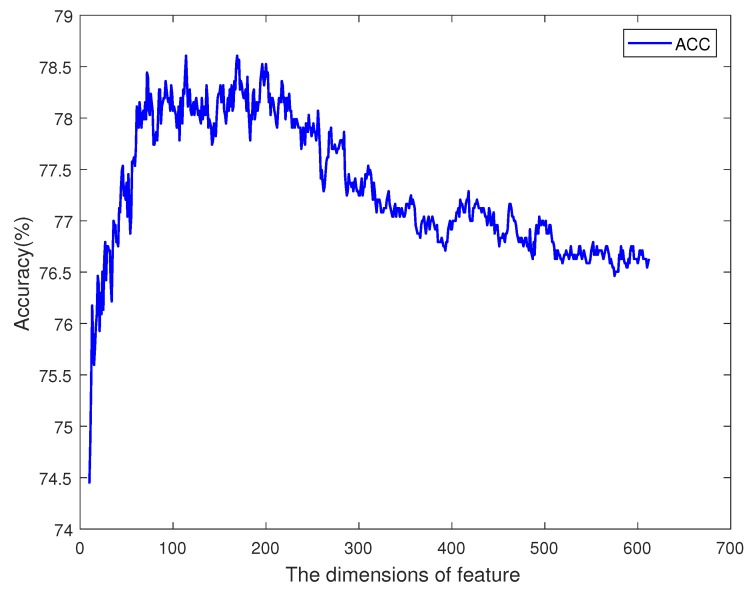
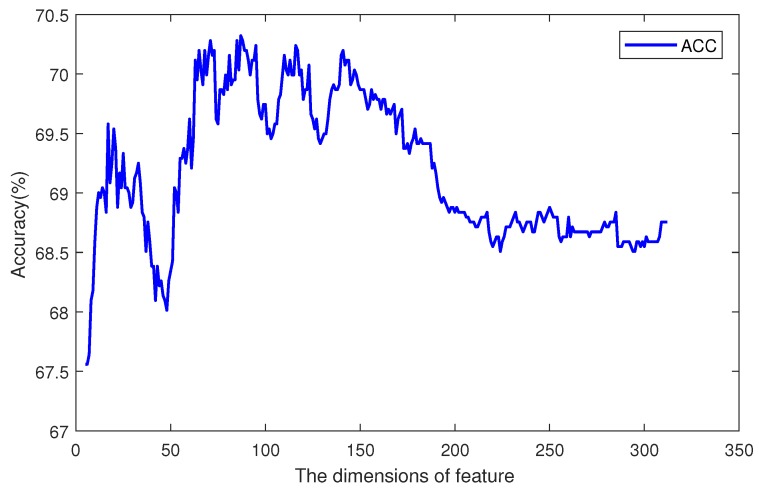
DNA methylation is an important biochemical process, and it has a close connection with many types of cancer. Research about DNA methylation can help us to understand the regulation mechanism and epigenetic reprogramming. Therefore, it becomes very important to recognize the methylation sites in the DNA sequence. In the past several decades, many computational methods-especially machine learning methods-have been developed since the high-throughout sequencing technology became widely used in research and industry. In order to accurately identify whether or not a nucleotide residue is methylated under the specific DNA sequence context, we propose a novel method that overcomes the shortcomings of previous methods for predicting methylation sites. We use -gram, multivariate mutual information, discrete wavelet transform, and pseudo amino acid composition to extract features, and train a sparse Bayesian learning model to do DNA methylation prediction. Five criteria-area under the receiver operating characteristic curve (AUC), Matthew's correlation coefficient (MCC), accuracy (ACC), sensitivity (SN), and specificity-are used to evaluate the prediction results of our method. On the benchmark dataset, we could reach 0.8632 on AUC, 0.8017 on ACC, 0.5558 on MCC, and 0.7268 on SN. Additionally, the best results on two scBS-seq profiled mouse embryonic stem cells datasets were 0.8896 and 0.9511 by AUC, respectively. When compared with other outstanding methods, our method surpassed them on the accuracy of prediction. The improvement of AUC by our method compared to other methods was at least 0.0399 . For the convenience of other researchers, our code has been uploaded to a file hosting service, and can be downloaded from: https://figshare.com/s/0697b692d802861282d3.
DNA 甲基化是一个重要的生化过程,它与许多类型的癌症密切相关。研究 DNA 甲基化可以帮助我们了解调控机制和表观遗传重编程。因此,识别 DNA 序列中的甲基化位点变得非常重要。在过去的几十年中,随着高通量测序技术在研究和工业中的广泛应用,许多计算方法——特别是机器学习方法——已经被开发出来。为了准确识别特定 DNA 序列背景下核苷酸残基是否被甲基化,我们提出了一种新的方法,克服了以前用于预测甲基化位点的方法的缺点。我们使用 -gram、多元互信息、离散小波变换和伪氨基酸组成来提取特征,并训练稀疏贝叶斯学习模型来进行 DNA 甲基化预测。使用五个标准——接受者操作特征曲线下的面积 (AUC)、马修相关系数 (MCC)、准确性 (ACC)、敏感性 (SN) 和特异性 (SP)——来评估我们方法的预测结果。在基准数据集上,我们在 AUC 上达到了 0.8632,在 ACC 上达到了 0.8017,在 MCC 上达到了 0.5558,在 SN 上达到了 0.7268。此外,在两个 scBS-seq 分析的小鼠胚胎干细胞数据集上,AUC 的最佳结果分别为 0.8896 和 0.9511。与其他优秀方法相比,我们的方法在预测精度上优于它们。与其他方法相比,我们的方法在 AUC 上的提高至少为 0.0399。为了方便其他研究人员,我们的代码已经上传到文件托管服务,可以从以下网址下载:https://figshare.com/s/0697b692d802861282d3。