Schroeder Jeremy W, Hirst William G, Szewczyk Gabriella A, Simmons Lyle A
Department of Molecular, Cellular and Developmental Biology, University of Michigan, Ann Arbor, MI 48109, USA.
Department of Molecular, Cellular and Developmental Biology, University of Michigan, Ann Arbor, MI 48109, USA.
Curr Biol. 2016 Mar 7;26(5):692-7. doi: 10.1016/j.cub.2016.01.016. Epub 2016 Feb 25.
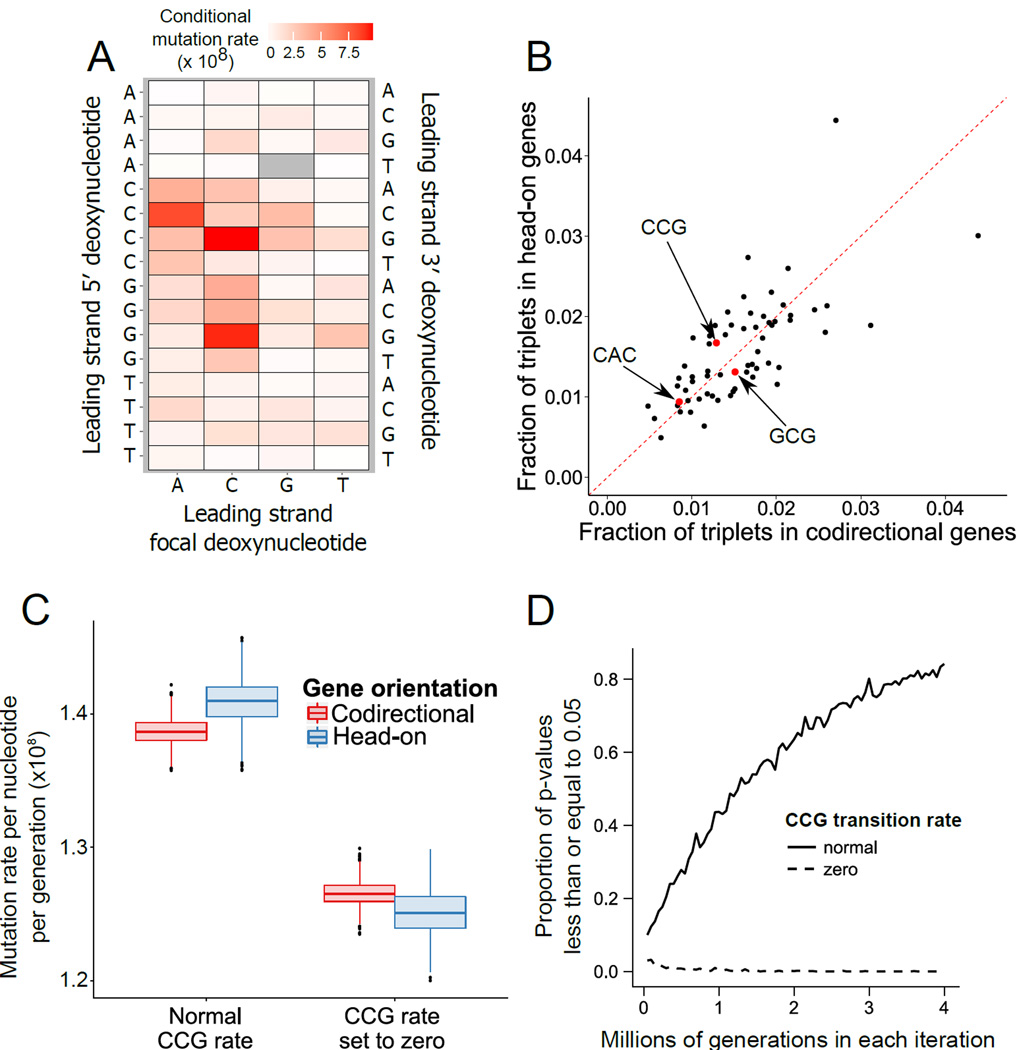
All organisms must replicate their genetic information accurately to ensure its faithful transmission. DNA polymerase errors provide an important source of genetic variation that can drive evolution. Understanding the origins of genetic variation will inform our understanding of evolution and the development of genetic diseases. A number of factors have been proposed to influence mutagenesis [1-10]. Here, we used mutation accumulation lines, whole-genome sequencing, and whole-transcriptome analysis to study the locations and rate at which mutations arise in bacteria with as little selection bias as possible [11, 12]. Our analysis of greater than 7,000 replication errors in over 180 sequenced lines that underwent a total of more than 370,000 generations has provided new insights into how DNA polymerase errors sculpt genetic variation and drive evolution. Homopolymer run enrichment outside of genes causes insertions and deletions in these regions. Genes encoded in the lagging strand are transcribed such that RNA polymerase and DNA polymerase collide head-on. Head-on genes have been proposed to mutate at a higher rate than genes transcribed codirectionally with DNA polymerase progression due to conflicts between transcription and DNA replication [6, 10]. We did not detect associations between the number of base pair substitutions in genes and their orientation or expression. Strikingly, any higher mutation rate for head-on genes can be explained by differing sequence composition between the leading and lagging strands and the error bias for DNA polymerase in specific sequence contexts. Therefore, we find local sequence context is the major determinant of mutagenesis in bacteria.
所有生物都必须准确复制其遗传信息,以确保其忠实传递。DNA聚合酶错误提供了遗传变异的一个重要来源,这种变异可以推动进化。了解遗传变异的起源将有助于我们理解进化和遗传疾病的发展。已经提出了许多因素来影响诱变作用[1-10]。在这里,我们使用突变积累系、全基因组测序和全转录组分析,尽可能减少选择偏差,来研究细菌中突变发生的位置和速率[11,12]。我们对超过180个测序系中超过7000个复制错误进行了分析,这些测序系总共经历了超过370000代,这为DNA聚合酶错误如何塑造遗传变异和推动进化提供了新的见解。基因外的同聚物重复富集导致这些区域的插入和缺失。滞后链中编码的基因转录时,RNA聚合酶和DNA聚合酶会迎头碰撞。由于转录和DNA复制之间的冲突,有人提出迎头基因的突变率高于与DNA聚合酶同向转录的基因[6,10]。我们没有检测到基因中碱基对替换的数量与其方向或表达之间的关联。引人注目的是,迎头基因任何较高的突变率都可以通过前导链和滞后链之间不同的序列组成以及特定序列背景下DNA聚合酶的错误偏差来解释。因此,我们发现局部序列背景是细菌诱变作用的主要决定因素。