Pignatelli Miguel, Vilella Albert J, Muffato Matthieu, Gordon Leo, White Simon, Flicek Paul, Herrero Javier
European Molecular Biology Laboratory, European Bioinformatics Institute
European Molecular Biology Laboratory, European Bioinformatics Institute.
Database (Oxford). 2016 Mar 15;2016. doi: 10.1093/database/bav127. Print 2016.
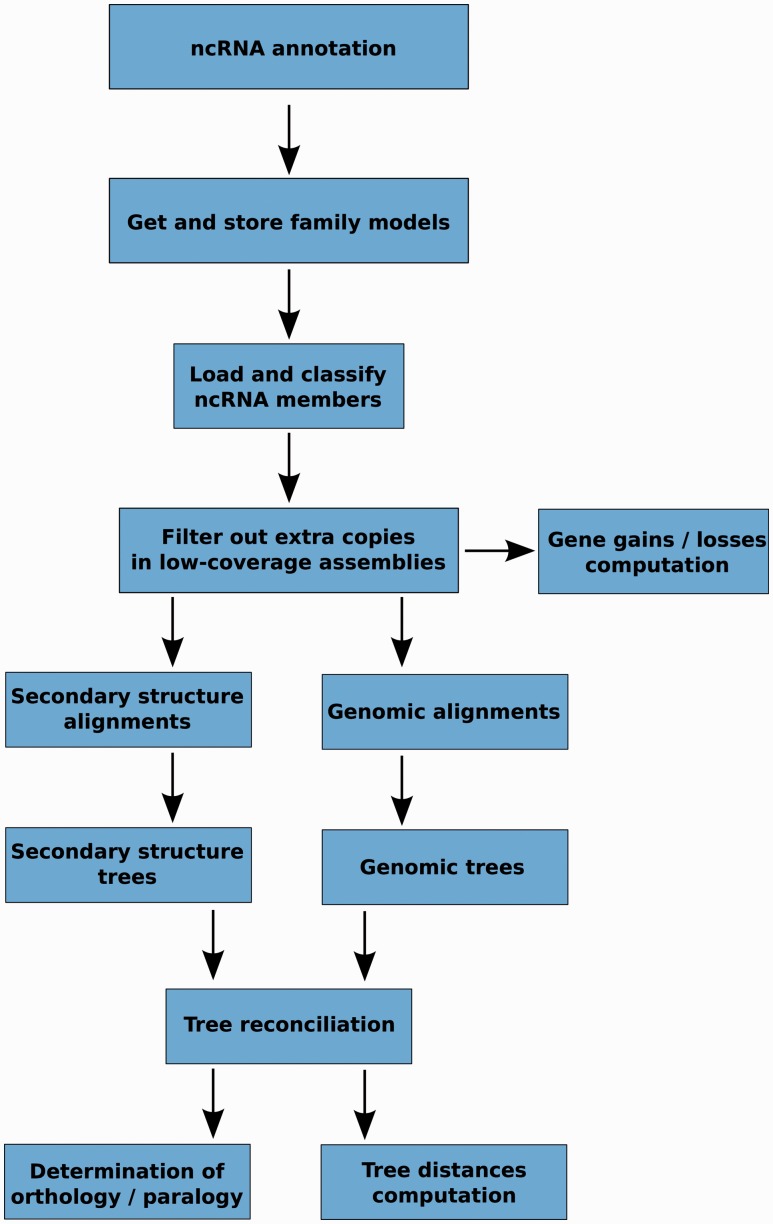
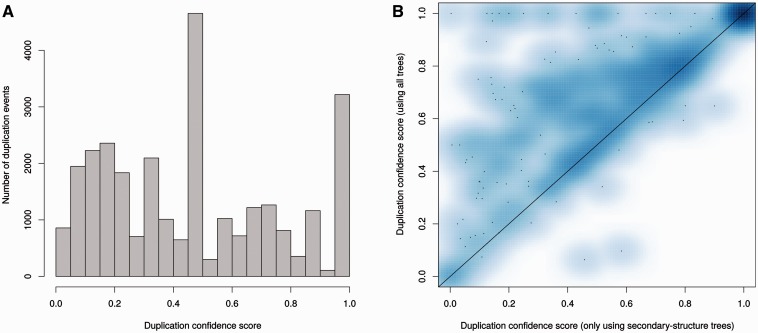
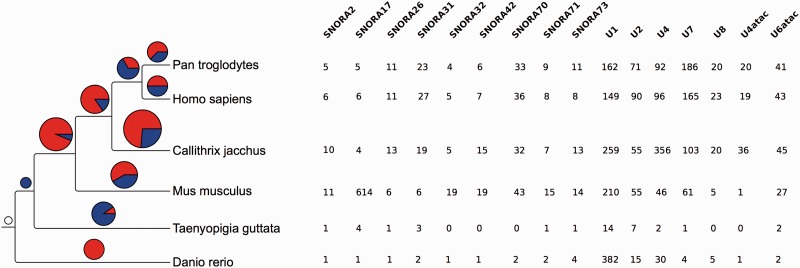
Annotation of orthologous and paralogous genes is necessary for many aspects of evolutionary analysis. Methods to infer these homology relationships have traditionally focused on protein-coding genes and evolutionary models used by these methods normally assume the positions in the protein evolve independently. However, as our appreciation for the roles of non-coding RNA genes has increased, consistently annotated sets of orthologous and paralogous ncRNA genes are increasingly needed. At the same time, methods such as PHASE or RAxML have implemented substitution models that consider pairs of sites to enable proper modelling of the loops and other features of RNA secondary structure. Here, we present a comprehensive analysis pipeline for the automatic detection of orthologues and paralogues for ncRNA genes. We focus on gene families represented in Rfam and for which a specific covariance model is provided. For each family ncRNA genes found in all Ensembl species are aligned using Infernal, and several trees are built using different substitution models. In parallel, a genomic alignment that includes the ncRNA genes and their flanking sequence regions is built with PRANK. This alignment is used to create two additional phylogenetic trees using the neighbour-joining (NJ) and maximum-likelihood (ML) methods. The trees arising from both the ncRNA and genomic alignments are merged using TreeBeST, which reconciles them with the species tree in order to identify speciation and duplication events. The final tree is used to infer the orthologues and paralogues following Fitch's definition. We also determine gene gain and loss events for each family using CAFE. All data are accessible through the Ensembl Comparative Genomics ('Compara') API, on our FTP site and are fully integrated in the Ensembl genome browser, where they can be accessed in a user-friendly manner. Database URL: http://www.ensembl.org.
直系同源基因和旁系同源基因的注释对于进化分析的许多方面都很有必要。传统上,推断这些同源关系的方法主要集中在蛋白质编码基因上,并且这些方法所使用的进化模型通常假定蛋白质中的位置是独立进化的。然而,随着我们对非编码RNA基因作用的认识不断增加,对经过一致注释的直系同源和旁系同源非编码RNA基因集的需求也日益增长。与此同时,诸如PHASE或RAxML等方法已经实现了考虑位点对的替代模型,以便对RNA二级结构的环和其他特征进行适当建模。在这里,我们提出了一个用于自动检测非编码RNA基因直系同源物和旁系同源物的综合分析流程。我们专注于Rfam中所代表的基因家族,并且为其提供了特定的协方差模型。对于在所有Ensembl物种中发现的每个家族的非编码RNA基因,使用Infernal进行比对,并使用不同的替代模型构建多棵树。同时,使用PRANK构建一个包含非编码RNA基因及其侧翼序列区域的基因组比对。该比对用于使用邻接法(NJ)和最大似然法(ML)创建另外两棵系统发育树。使用TreeBeST合并来自非编码RNA和基因组比对的树,将它们与物种树进行协调,以识别物种形成和复制事件。最终的树用于根据菲奇的定义推断直系同源物和旁系同源物。我们还使用CAFE确定每个家族的基因获得和丢失事件。所有数据都可以通过Ensembl比较基因组学(“Compara”)应用程序编程接口、我们的FTP站点访问,并且完全集成在Ensembl基因组浏览器中,在那里可以以用户友好的方式进行访问。数据库网址:http://www.ensembl.org。