Lee Inkyu S, Carvalho Claudia M B, Douvaras Panagiotis, Ho Seok-Man, Hartley Brigham J, Zuccherato Luciana W, Ladran Ian G, Siegel Arthur J, McCarthy Shane, Malhotra Dheeraj, Sebat Jonathan, Rapoport Judith, Fossati Valentina, Lupski James R, Levy Deborah L, Brennand Kristen J
Departments of Psychiatry and Neuroscience, Icahn School of Medicine at Mount Sinai, 1425 Madison Avenue, New York, NY 10029.
Baylor College of Medicine, Department of Molecular and Human Genetics, One Baylor Plaza, Houston, TX 77030.
NPJ Schizophr. 2015 Jun 24;1:15019-. doi: 10.1038/npjschz.2015.19.
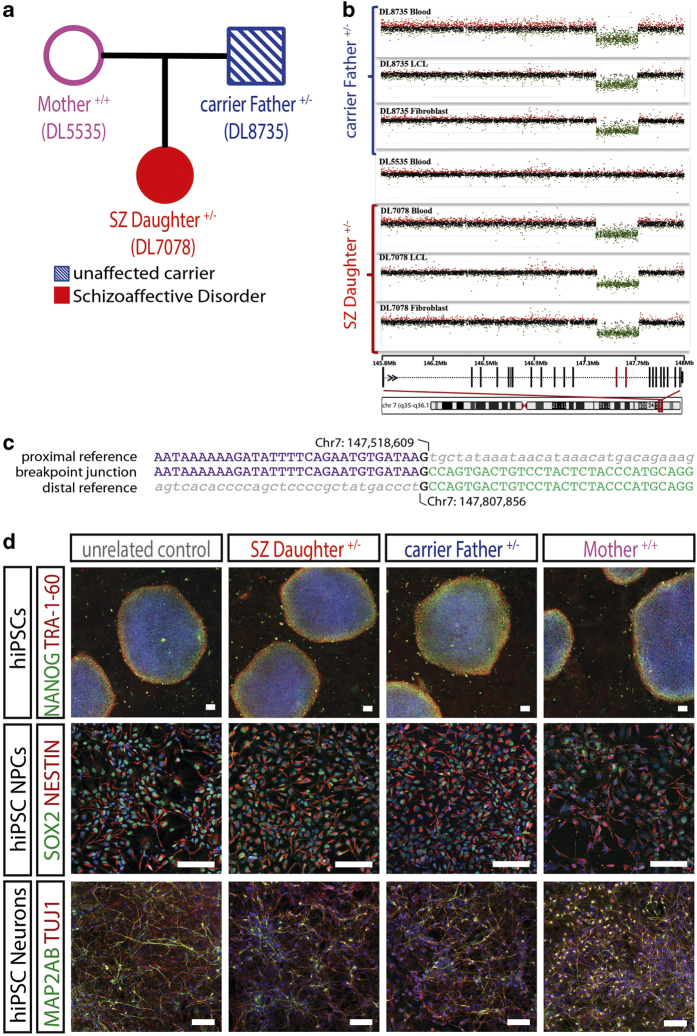
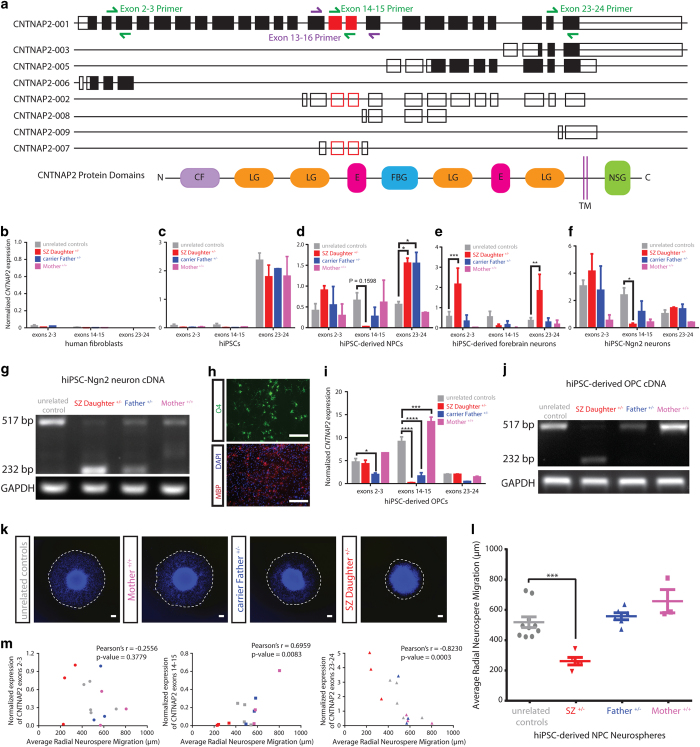
Neurodevelopmental disorders, such as autism spectrum disorders (ASD) and schizophrenia (SZ), are complex disorders with a high degree of heritability. Genetic studies have identified several candidate genes associated with these disorders, including contactin-associated protein-like 2 (). Traditionally, in animal models or , the function of has been studied by genetic deletion or transcriptional knockdown, which reduce the expression of the entire gene; however, it remains unclear whether the mutations identified in clinical settings are sufficient to alter expression in human neurons. Here, using human induced pluripotent stem cells (hiPSCs) derived from two individuals with a large (289kb) and heterozygous deletion in (affecting exons 14-15) and discordant clinical outcomes, we have characterized expression patterns in hiPSC neural progenitor cells (NPCs), two independent populations of hiPSC-derived neurons and hiPSC-derived oligodendrocyte precursor cells (OPCs). First, we observed exon-specific changes in expression in both carriers; although the expression of exons 14-15 is significantly decreased, the expression of other exons is upregulated. Second, we observed significant differences in patterns of allele-specific expression in carriers that were consistent with clinical outcome. Third, we observed a robust neural migration phenotype that correlated with diagnosis and exon- and allele-specific expression patterns, but not with genotype. In all, our data highlight the importance of considering the nature, location and regulation of mutated alleles when attempting to connect GWAS studies to gene function.
神经发育障碍,如自闭症谱系障碍(ASD)和精神分裂症(SZ),是具有高度遗传性的复杂疾病。基因研究已经确定了几个与这些疾病相关的候选基因,包括接触蛋白相关蛋白样2()。传统上,在动物模型或中,通过基因缺失或转录敲低来研究的功能,这会降低整个基因的表达;然而,临床环境中鉴定出的突变是否足以改变人类神经元中的表达仍不清楚。在这里,我们使用来自两名携带大的(289kb)杂合缺失(影响外显子14 - 15)且临床结果不一致的个体的人类诱导多能干细胞(hiPSC),对hiPSC神经祖细胞(NPC)、两个独立的hiPSC衍生神经元群体和hiPSC衍生少突胶质细胞前体细胞(OPC)中的表达模式进行了表征。首先,我们在两名携带者中均观察到了外显子特异性的表达变化;虽然外显子14 - 15的表达显著降低,但其他外显子的表达上调。其次,我们在携带者中观察到等位基因特异性表达模式的显著差异,这些差异与临床结果一致。第三,我们观察到一种与诊断以及外显子和等位基因特异性表达模式相关,但与基因型无关的强大神经迁移表型。总之,我们的数据突出了在尝试将全基因组关联研究(GWAS)与基因功能联系起来时,考虑突变等位基因的性质、位置和调控的重要性。