Su Zhipeng, Zhu Jiawen, Xu Zhuofei, Xiao Ran, Zhou Rui, Li Lu, Chen Huanchun
State Key Laboratory of Agricultural Microbiology, College of Veterinary Medicine, Huazhong Agricultural University, Wuhan 430070, China.
Cooperative Innovation Center of Sustainable Pig Production, Wuhan 430070, China.
PLoS One. 2016 Mar 28;11(3):e0152363. doi: 10.1371/journal.pone.0152363. eCollection 2016.
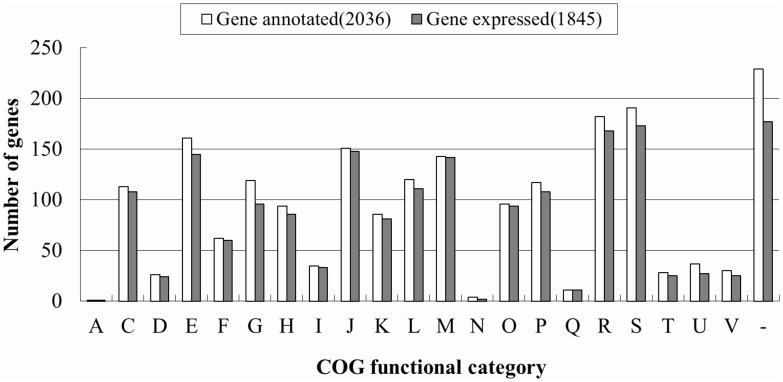
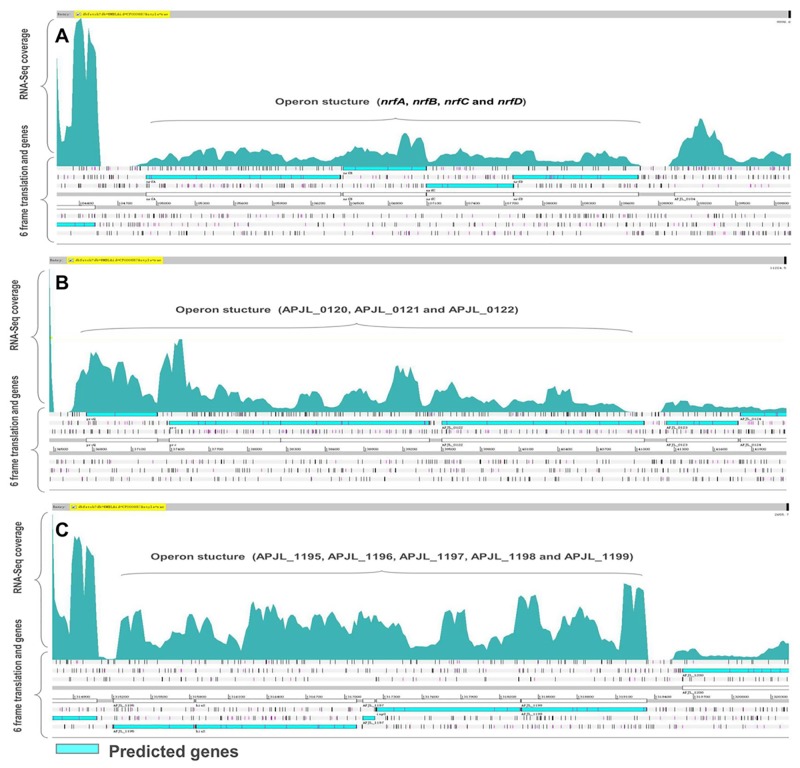
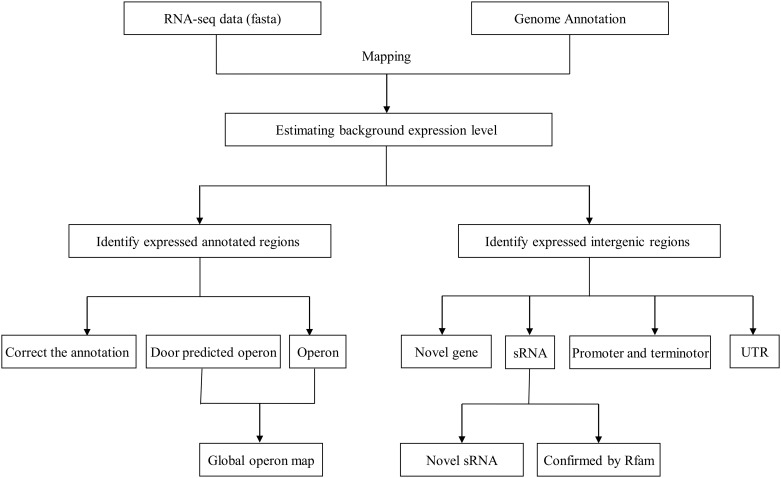
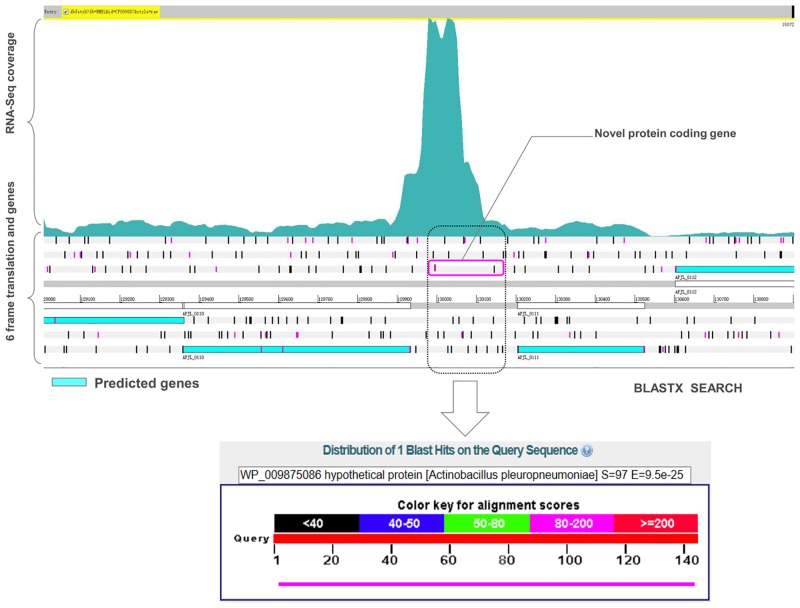
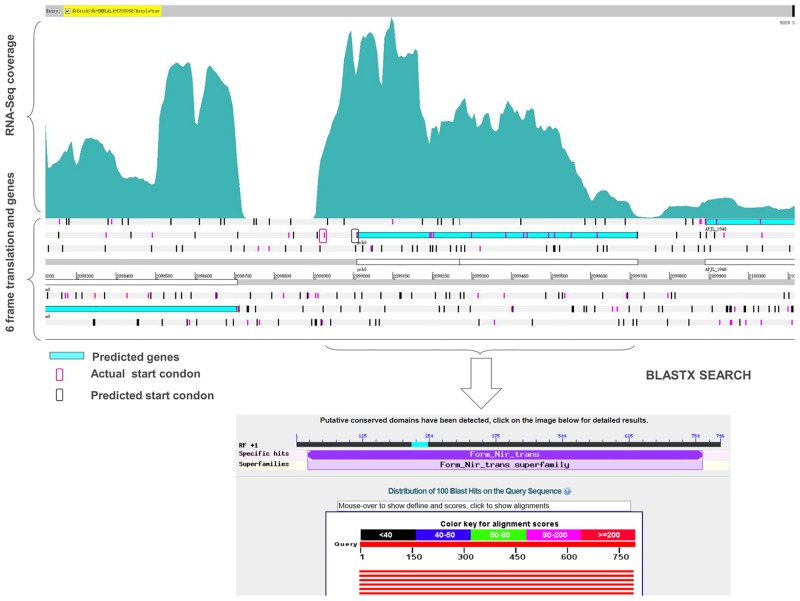
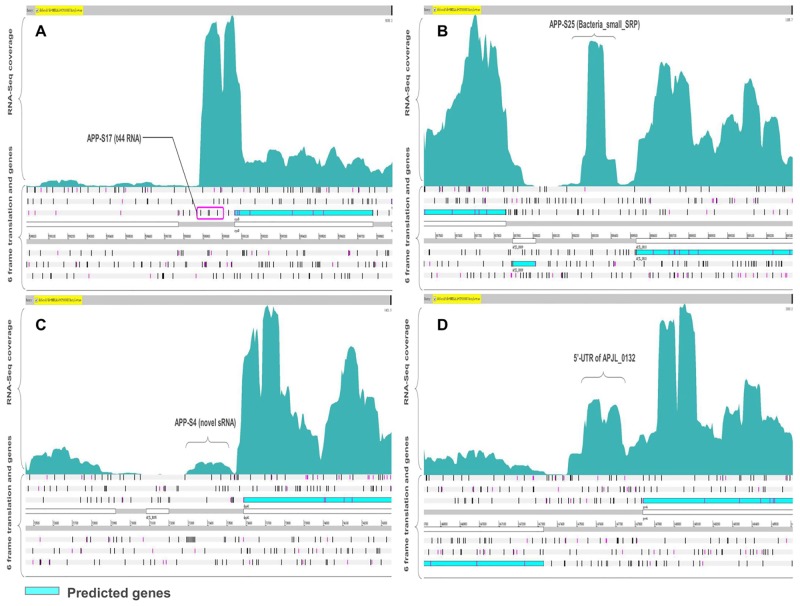
Actinobacillus pleuropneumoniae is the pathogen of porcine contagious pleuropneumoniae, a highly contagious respiratory disease of swine. Although the genome of A. pleuropneumoniae was sequenced several years ago, limited information is available on the genome-wide transcriptional analysis to accurately annotate the gene structures and regulatory elements. High-throughput RNA sequencing (RNA-seq) has been applied to study the transcriptional landscape of bacteria, which can efficiently and accurately identify gene expression regions and unknown transcriptional units, especially small non-coding RNAs (sRNAs), UTRs and regulatory regions. The aim of this study is to comprehensively analyze the transcriptome of A. pleuropneumoniae by RNA-seq in order to improve the existing genome annotation and promote our understanding of A. pleuropneumoniae gene structures and RNA-based regulation. In this study, we utilized RNA-seq to construct a single nucleotide resolution transcriptome map of A. pleuropneumoniae. More than 3.8 million high-quality reads (average length ~90 bp) from a cDNA library were generated and aligned to the reference genome. We identified 32 open reading frames encoding novel proteins that were mis-annotated in the previous genome annotations. The start sites for 35 genes based on the current genome annotation were corrected. Furthermore, 51 sRNAs in the A. pleuropneumoniae genome were discovered, of which 40 sRNAs were never reported in previous studies. The transcriptome map also enabled visualization of 5'- and 3'-UTR regions, in which contained 11 sRNAs. In addition, 351 operons covering 1230 genes throughout the whole genome were identified. The RNA-Seq based transcriptome map validated annotated genes and corrected annotations of open reading frames in the genome, and led to the identification of many functional elements (e.g. regions encoding novel proteins, non-coding sRNAs and operon structures). The transcriptional units described in this study provide a foundation for future studies concerning the gene functions and the transcriptional regulatory architectures of this pathogen.
胸膜肺炎放线杆菌是猪传染性胸膜肺炎的病原体,猪传染性胸膜肺炎是一种猪的高度传染性呼吸道疾病。尽管胸膜肺炎放线杆菌的基因组在几年前就已测序,但关于全基因组转录分析以准确注释基因结构和调控元件的信息有限。高通量RNA测序(RNA-seq)已被应用于研究细菌的转录图谱,它可以高效、准确地识别基因表达区域和未知转录单元,特别是小非编码RNA(sRNA)、非翻译区(UTR)和调控区域。本研究的目的是通过RNA-seq全面分析胸膜肺炎放线杆菌的转录组,以改进现有的基因组注释,并增进我们对胸膜肺炎放线杆菌基因结构和基于RNA的调控的理解。在本研究中,我们利用RNA-seq构建了胸膜肺炎放线杆菌的单核苷酸分辨率转录组图谱。从一个cDNA文库中产生了超过380万个高质量读数(平均长度约90 bp),并与参考基因组进行比对。我们鉴定出32个编码新蛋白的开放阅读框,这些开放阅读框在先前的基因组注释中被错误注释。基于当前基因组注释的35个基因的起始位点得到了校正。此外,在胸膜肺炎放线杆菌基因组中发现了51个sRNA,其中40个sRNA在先前的研究中从未被报道过。转录组图谱还使得5'-和3'-UTR区域得以可视化,其中包含11个sRNA。此外,在整个基因组中鉴定出351个操纵子,覆盖1230个基因。基于RNA-Seq的转录组图谱验证了注释基因并校正了基因组中开放阅读框的注释,并导致鉴定出许多功能元件(例如编码新蛋白的区域、非编码sRNA和操纵子结构)。本研究中描述的转录单元为今后关于该病原体的基因功能和转录调控结构的研究提供了基础。